Hematology & Oncology


Hematology & Oncology
The Hematology & Oncology department at Labor Berlin was created in 2010/2011 by merging the hematology-oncology diagnostic laboratories of the two Charité sites Campus Benjamin Franklin and Campus Virchow-Klinikum with the hematology special diagnostics department of the Vivantes clinics.
Diagnostics in hemato-oncology
Alongside cardiovascular diseases, cancer is one of the leading causes of death and also represents a major health policy challenge. In hardly any other sub-discipline of medicine have the findings of basic research in the fields of immunology and genetics found such direct application as in hemato-oncology. In this field, for example, the first therapeutic monoclonal antibodies were used and the first molecularly targeted therapies were developed. In no other field of modern medicine is genetic-immunological diagnostics as refined as in acute leukemia, for example.
Tumor diseases are usually extremely heterogeneous. A comprehensive set of diagnostic tools is therefore often necessary to make an accurate diagnosis. Even today – although some of the basic principles are more than 100 years old – cytomorphology is still a cornerstone of hematological diagnostics. In addition, there are immunological methods such as flow cytometry (immunophenotyping) and genetic methods such as cytogenetics and molecular genetics. In many hematologic diseases/neoplasms, a reliable diagnosis can only be made by looking at all these diagnostic findings together.
In addition, some diagnostic features (immunological or genetic) have a prognostic implication. As part of large therapy studies, for example, high- and low-risk features have been characterized for many haematological diseases. The identification of these characteristics is often important, as therapy can be guided by them.
Diagnostics
-
Cytomorphology is also a cornerstone of diagnostics in modern hematology. Depending on the problem, the morphological findings are supplemented by immunocytology (flow cytometry), cytogenetics and molecular biology.
In addition to manual differential blood counts, the cytology laboratory primarily prepares bone marrow smears and cytological centrifuge preparations from cerebrospinal fluid and effusion fluids (pleural effusion, ascites, etc.). In addition to conventional cytological assessment, cytochemical and immunocytological methods are also used. The aim with all submissions is to prepare the findings quickly: if the sample is received on time and a particular urgency is communicated, even on the same day!
These examinations are offered:
- Manual differential blood count, if necessary after leukocyte enrichment
- Bone marrow cytology, if necessary including cytochemistry, iron staining and immunocytology
- Cytology from effusions (e.g. pleural effusion, ascites), including immunocytology if necessary
- Cerebrospinal fluid cytology, including immunocytology if necessary
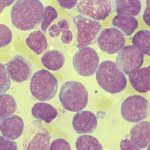
Bone marrow exchange for acute myeloid leukemia (AML)
Immunocytological staining: carcinoma cells in the pleural effusion
The samples are processed and the findings prepared on working days from Monday to Friday. EDTA (or citrate) must be used as an anticoagulant. The quality of the findings and their interpretation depend largely on the age and quality of the sample material as well as on accompanying clinical data. Fast transportation routes are therefore recommended. The important clinical findings should be transmitted in full. A current blood count with differential blood count is particularly important for bone marrow cytology. The laboratory team will be happy to discuss the findings by telephone.
Preanalytical notes – Cytology
The anticoagulant for all cytological examinations is EDTA. Citrate is also possible in exceptional cases. Heparin, on the other hand, is unfavorable and should not be used.
Suitable test materials are
- Bone marrow aspirate (5 ml)
- Peripheral blood (routine EDTA blood tubes)
- Effusion material (ascites, pleural effusion, pericardial effusion, etc.)
- Liquor
Care should be taken to ensure that the transportation time does not exceed 24 hours and that the material arrives at the laboratory before weekends by Friday, 14:00 at the latest. In principle, material should be sent with an express service (overnight service) to avoid excessively long transportation times.
-
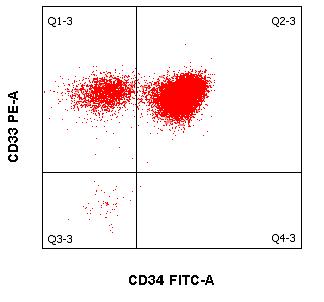
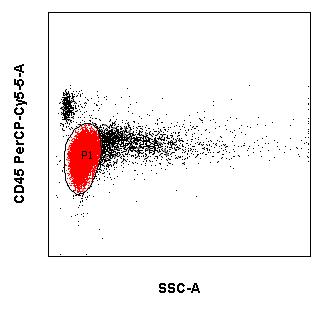
In addition to morphology, cytogenetics and molecular biology, immunocytological diagnostics (using flow cytometry) is an important cornerstone in the diagnosis and assessment of leukemias and lymphomas. Immunophenotyping of malignant cells is usually performed in peripheral blood and bone marrow. However, body fluids such as cerebrospinal fluid and effusions can also be sent as examination material. Modern multiparametric flow cytometry is essentially based on advances in three areas:
- Laser optics
- Computer-aided data processing
- Development of new fluorescent dyes for coupling to the corresponding monoclonal antibodies
When several fluorescent dye-coupled antibodies are combined and the different scattering light properties of cells are utilized, it is possible:
- classify malignant hematologic neoplasms and
- to assess the success of therapy in the context of a remission assessment and the control of minimal residual disease (MRD).
The aim for all submissions is a rapid preparation of findings: if samples are received on time, they are sent by fax on the same day.
The following tests are carried out in the laboratory:
- Immunophenotyping of leukemias and lymphomas (5-10 ml EDTA blood or -KM)
- Remission assessment incl. Detection of MRD (minimal residual disease) in leukemias and lymphomas (5-10 ml EDTA blood or -KM)
- Detection of leukemia/lymphoma cells in cerebrospinal fluid and effusion fluids (5 ml sample material anticoagulated with EDTA)
- Detection of myeloma cells (5ml EDTA blood or KM)
- PNH diagnostics (10 ml EDTA blood)
- Quantification of CD34+ hematopoietic stem cells (2 ml of EDTA blood or stem cell aspirate)
-
EMA test for spherocytosis (2 ml EDTA blood)
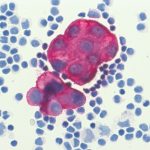
Hairy cell leukemia (peripheral blood)
Acute myeloid leukemia (bone marrow)
The samples are processed and the findings prepared on working days from Monday to Friday. The quality of the findings and their interpretation depend largely on the age and quality of the sample material as well as on accompanying clinical information. Fast transportation routes are therefore recommended. The important clinical findings incl. Blood count and differential blood count should be submitted in full. The laboratory team will be happy to discuss the findings over the phone.
Preanalytical information – Flow cytometry
The anticoagulant for all flow cytometric tests is always EDTA. Heparin is less suitable and should only be used if nothing else is available (e.g. accidental collection in the wrong syringe during a bone marrow puncture).
Suitable test materials are
- Bone marrow aspirate (5 – 10 ml)
- Peripheral blood (5 – 10 ml)
- Effusion material (ascites, pleural effusion, pericardial effusion, etc.), if possible 10 ml
- Liquor
- PNH diagnosis from peripheral blood !
Samples should always be shipped using an express service (overnight service) in order to keep transportation times as short as possible. The samples should not be older than 24 hours when they arrive at the laboratory. Before weekends, please ensure that the sample arrives at the laboratory by noon on Friday at the latest.
-
Molecular tumor genetics deals with the analysis of molecular changes in malignant diseases. Knowledge of the molecular changes in malignant diseases has grown exponentially in recent decades, especially since the first complete sequencing of the human genome in 2001 as part of the Human Genome Project. Accordingly, the diagnostic possibilities have also expanded considerably.
The most important diagnostic methods in the field of molecular tumor genetics are the various polymerase chain reaction (PCR) techniques and DNA sequencing. Molecular diagnostic tumor genetics has two main fields of application:
- The detection of molecular changes in tumors. These include chromosome translocations or mutations in the broadest sense. In some chromosome translocations, chimeric fusion genes are formed that are transcribed as mRNA. This chimeric mRNA can be detected by RT-PCR. Point mutations can lead to amino acid substitutions and thus to functional changes in genes. Other mutations lead to reading frame shifts, loss of splice sites, etc. and thus to a loss of function of the gene. many of these molecular changes in tumors are of prognostic or therapeutic significance.
- Progression studies using molecular markers. Many hematological diseases are characterized by molecular changes that can be quantified using quantitative PCR. By regularly quantifying these changes during the course of the disease, valuable information can be obtained about the extent of the disease, e.g. the effectiveness of a therapy can be assessed. These measurements are particularly important for acute and chronic leukemias (MRD = minimal residual disease).
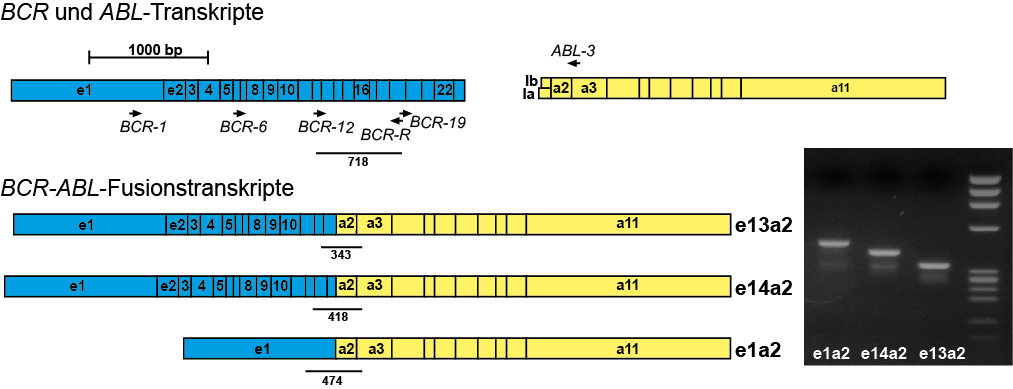
Example: Detection of the BCR-ABL fusion using RT-PCR
Range of methods
The most important molecular genetic methods used in the Berlin laboratory are:
- Conventional PCR and RT-PCR (detection in agarose gel)
- Long-distance PCR
- real-time quantitative PCR (genomic DNA or cDNA)
- DNA sequencing (Sanger and next-generation sequencing NGS)
- Chimerism analyses after allogeneic transplantation
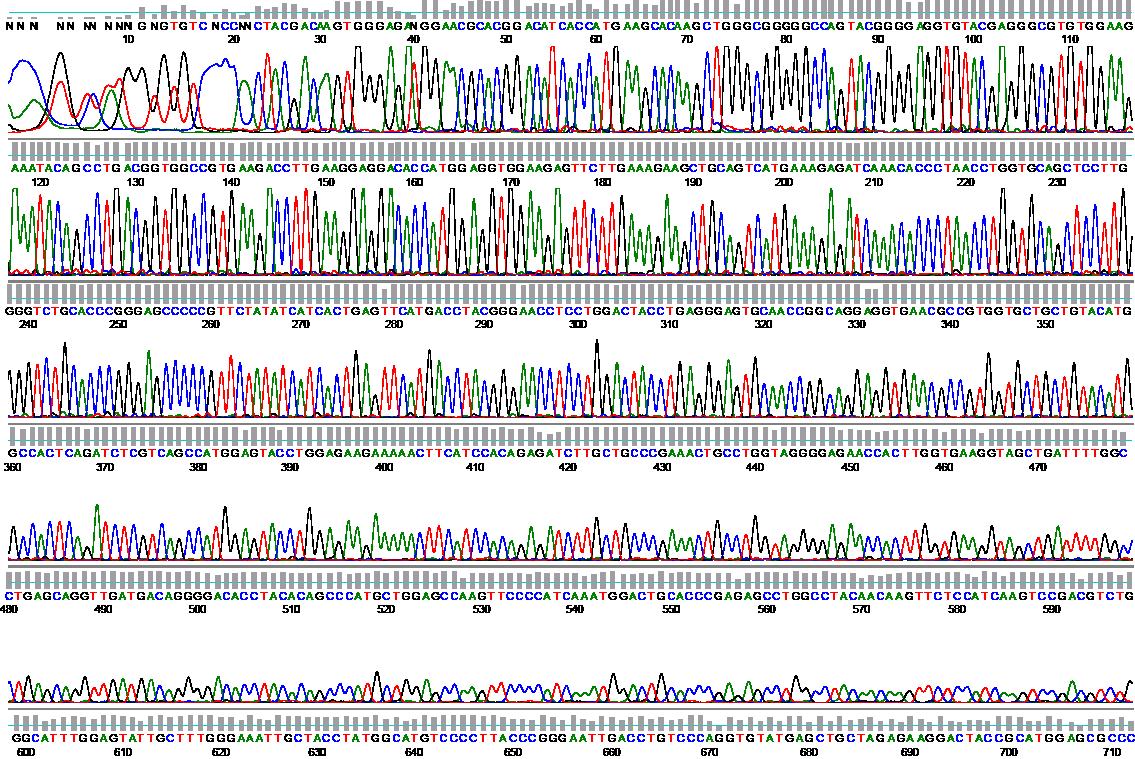
Example: BCR-ABL mutation analysis using sequencing
Preanalytical notes
The anticoagulant for all molecular genetic tests is EDTA. Citrate can also be used in exceptional cases. Heparin, on the other hand, is unfavorable (inhibits enzymatic reactions such as PCR) and should only be used if nothing else is available (e.g. accidental collection in the wrong syringe during a bone marrow puncture).
In principle, the following can be used as test material:
- Bone marrow aspirate (5 ml)
- Peripheral blood (10 ml)
- Effusion material (ascites, pleural effusion)
- Liquor
- Lymph node or tumor biopsy (always unfixed, i.e. in NaCl or water!)
If tumor material or lymph nodes are sent in, please contact us in advance by telephone. Molecular genetic diagnostics from cerebrospinal fluid usually only has a chance of success if it is very rich in cells. Here too, prior consultation by telephone is recommended.
For chimerism analyses after allogeneic transplantation, it must be ensured that our laboratory has or had material from both the donor and the recipient prior to transplantation. The prerequisite for a chimerism analysis is knowledge of the differences in the chimerism markers between donor and recipient. The chimerism analysis can either be “unsorted” (“total chimerism”) from total blood, bone marrow, etc., or “sorted” (“sorted chimerism”), i.e. from isolated cell fractions, e.g. CD34+ stem cells. If a “sorted chimerism” is requested, the material must be fresh, i.e. the cells to be sorted out must still be intact.
Samples should be sent by express mail, especially for time-critical samples (acute leukemia). If possible, shipping should not take place on a Friday.
-
In tumor cytogenetics, acquired chromosomal abnormalities in hematological neoplasms and solid tumors are examined. Characteristic chromosomal abnormalities that frequently occur in a certain type of tumor are associated with neoplastic transformation: they are considered primary abnormalities. Secondary abnormalities occur as the disease progresses, when the genome becomes increasingly unstable. They can contribute to the progression of the tumor.
What can tumor cytogenetics do?
The cytogenetic examination of peripheral blood and bone marrow is now part of standard diagnostics for malignant hematologic diseases. It comprises classical chromosome analysis together with fluorescence in situ hybridization. Cytogenetic analysis makes an important contribution to confirming and specifying the suspected diagnosis, prognosis assessment and treatment options. It is also used to monitor the course of the disease. The identification of specific genetic markers of the malignant cell clone can also be used to monitor the course of therapy.Indications
Myeloid leukemias
- Myeloproliferative neoplasms (MPN)
- Myelodysplastic / myeloproliferative diseases
- Myelodysplastic syndromes (MDS)
- Acute myeloid leukemia (AML)
- Biphenotypic acute leukemia (BAL)
Acute lymphoblastic leukemia
- B-cell acute lymphoblastic leukemia (B-ALL)
- T-cell acute lymphoblastic leukemia (T-ALL)
- Non-Hodgkin’s lymphoma (NHL)
Methods
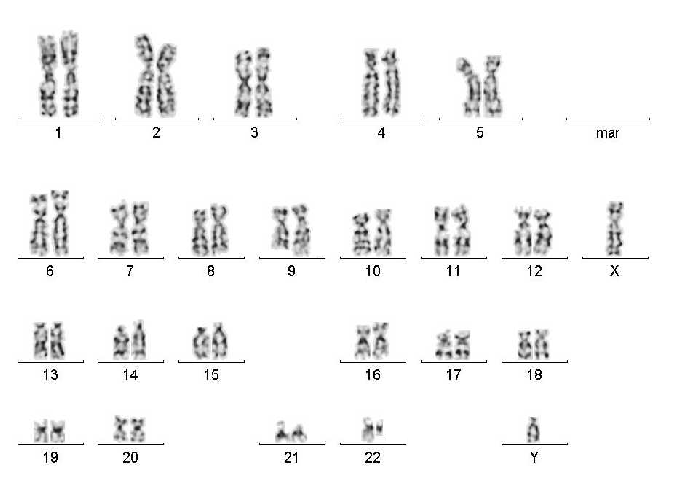
Karyogram
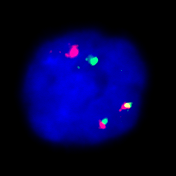
Fluorescence in situ hybridization
Chromosome analysis is performed on prepared metaphases using computer-assisted karyotyping.
FISH analysis allows even finer submicroscopic changes to be visualized. It also enables screening for frequent, tumor-typical aberrations in the case of insufficient proliferation behavior of the tumor cells (such as plasmocytoma or chronic lymphocytic leukemia).
Spectral karyotyping (SKY) is an important further development of the FISH method. It is becoming increasingly important in molecular cytogenetic tumor diagnostics. SKY can also be used to break down complex aberrant rearrangements with unknown translocation partners or marker chromosomes of unknown origin.
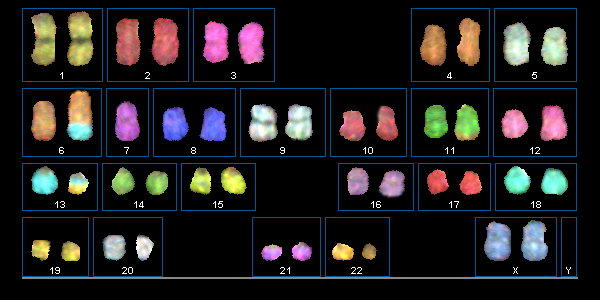
Spectral karyotyping
Preanalytical notes
Na-Heparin bone marrow 10ml / Peripheral blood 20ml
Disease entities
The subordinate pages contain some general information and diagnostic information on various hematologic diseases. The classification into disease groups is based on the WHO classification of hematologic neoplasms from 2008.
Each subchapter contains information on how to carry out the most rational diagnosis possible.
For example, the following chapters provide information on which genetic tests are useful and when (usually depending on the associated immunological and morphological findings).
- Acute myeloid leukemia
- Acute lymphoblastic leukemia
- Myelodysplastic syndromes
- Eosinophil syndromes and
- Non-Hodgkin’s lymphomas
Selected individual diseases are also discussed in more detail in the following chapters.
- Non-Hodgkin’s lymphomas
- Myelodysplastic syndromes
- MDS/MPN mixed forms and
- Myeloproliferative neoplasms
-
Basic laboratory diagnostics for MPN includes microscopic differential blood counts, determination of clinical-chemical parameters and bone marrow diagnostics.
Bone marrow diagnostics should include the following examinations at initial diagnosis:
- Bone marrow histology
- Bone marrow cytology
- Immunophenotyping
- Molecular genetics
- Cytogenetics
Bone marrow histology is the only one of the above-mentioned examinations that is not offered at the Berlin laboratory. It provides information about the degree of fibrosis in the bone marrow and is important in the differential diagnosis of myeloproliferative neoplasms.
Chronic myeloid leukemia (CML)
In bone marrow cytology, chronic myeloid leukemia usually shows an increase in all 3 cell series (megakaryopoiesis, myelopoiesis, erythropoiesis), with myelopoiesis clearly dominating, often associated with eosinophilia and basophilia.
The BCR-ABL fusion gene can almost always be detected molecularly by RT-PCR. If the molecular genetic analysis cannot detect this fusion gene, CML is unlikely. Occasionally, no translocation t(9;22) or Philadelphia chromosome can be detected by conventional cytogenetics, but the BCR-ABL fusion can be detected by molecular genetics. Molecular genetic analysis can also be performed on peripheral blood in cases of suspected CML. As there are many different transcript variants of BCR-ABL, molecular genetic analysis must be geared towards detecting all of them.The cytogenetic analysis should be carried out from the bone marrow and only if not otherwise possible from the peripheral blood, as the cells obtained from the peripheral blood can often only be insufficiently stimulated to divide in culture. Cytogenetically, CML typically shows the translocation t(9;22)(q34;q22), which often shows a shortened chromosome 22 (“Philadelphia chromosome”).
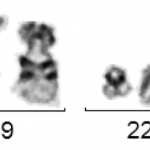
t(9;22)(q34;q11) with Philadelphia chromosome
However, the value of cytogenetic analysis lies not only in the detection of t(9;22), but also in the fact that other additional aberrations can be detected. Typical additional aberrations in CML are the loss of the long arm of chromosome 9 (del(9q)), an additional Philadelphia chromosome or a second t(9;22) and an inversion of chromosome 17 (inv(17)). All three additional aberrations are considered to be prognostically unfavorable, are found more frequently in the accelerated phase or the blast relapse and can be an indication of rapid progression of the disease. In addition, there are very rare cases in which the clinical picture is that of CML, but genetically there is no translocation t(9;22) or no BCR-ABL fusion gene, but a fusion of the BCR gene with other tyrosine kinases (e.g. BCR-FGFR3, BCR-JAK2, etc.). Cytogenetics should always be supplemented by fluorescence in situ hybridization (FISH) for the BCR-ABL fusion gene.
Follow-up examinations for CML
Follow-up examinations for BCR-ABL-positive CML are carried out using quantitative PCR (qPCR). If the disease level is high, additional cytogenetic (+FISH) examinations are also useful. Peripheral blood is sufficient as test material for molecular genetics, as it has been shown that the BCR-ABL level correlates relatively well between bone marrow and blood. The European Leukemia Net has defined certain time landmarks that should be reached during therapy with tyrosine kinase inhibitors. If these targets are not reached, this can be seen as a warning signal. In patients undergoing therapy with tyrosine kinase inhibitors, a qPCR from peripheral blood should be performed every 3 months.BCR-ABL mutation analyses
If there is a continuous significant increase in the BCR-ABL level(by more than one order of magnitude) in several consecutive qPCR tests during treatment with a tyrosine kinase inhibitor, a BCR-ABL mutation analysis should be performed. This is done by direct sequencing from the most recently submitted material with the highest BCR-ABL level. If a BCR-ABL mutation is found, this may be a valuable decision-making aid for switching to another tyrosine kinase inhibitor.BCR-ABL-negative myeloproliferative neoplasms (MPN)
The majority of MPNs do not have the BCR-ABL fusion gene. However, other molecular aberrations can often be detected.
MPN with JAK2 V617F mutation
Since the first description in 2005, it has been known that many MPNs are molecularly characterized by a point mutation in Janus kinase 2 (JAK2). This point mutation leads to the amino acid exchange valine -> phenylalanine at position 617 of the JAK2 protein (V617F mutation) and consecutively to the constitutional activation of the JAK2 kinase.The JAK2 V617F mutation is found in varying percentages in different MPNs – in more than 90% in polycythemia vera (PV), and in a significantly lower percentage in primary myelofibrosis (PMF) or essential thrombocythemia (ET). A negative JAK2 V617F result therefore does not rule out the diseases mentioned. JAK2 mutations have been included by the WHO as a major criterion for the diagnosis of PV, PMF or ET.
MPN with JAK2 exon 12 mutations
Mutations in other sections of the JAK2 gene are much rarer than the JAK2 V617F mutation, which is based on a nucleotide mutation in JAK2 exon 14. These are mainly found in exon 12 of the JAK2 gene. Patients with JAK2 exon 12 mutations almost always present with isolated erythrocytosis, i.e. polycythemia vera. These mutations are very rare and should not be routinely determined in every MPN diagnosis. The diagnosis is recommended in the case of a clinical picture of PV and simultaneously undetectable JAK2 V617F mutation.MPN with calreticulin (CALR) mutation
It has been known since 2013 that around 25 to 30 % of cases of PMF and ET have mutations in the last coding exon of the CALR gene. These mutations hardly ever occur in PV. The mutations lead to a reading frame shift of -1 nucleotide, with the result that the transcribed mutated calreticulin protein has an altered carboxy-terminal end. This results in the protein being incorrectly sorted into subcellular compartments. Mainly two recurrent mutation types are observed – “type 1” and “type 2”, but there are also numerous others. CALR mutations are largely considered to have a better prognosis than JAK2 mutations.MPN with MPL W515L mutation
In ET and PMF, a point mutation is occasionally found in the thrombopoietin receptor gene MPL, which leads to the amino acid exchange tryptophan for leucine at position 515 (W515L). The mutation is found in 5% or less of all patients with ET or PMF, and is more common in PMF than in ET. Detection of the MPL W515L mutation is one of the WHO major diagnostic criteria for PMF and ET.Systemic mastocytosis
In adult patients, systemic mastocytosis often has activating mutations in the tyrosine kinase KIT located on chromosome 4q11. By far the most common amino acid exchange is aspartate -> valine at position 816 (D816V).WHO diagnostic criteria for PV, ET and PMF from 2008
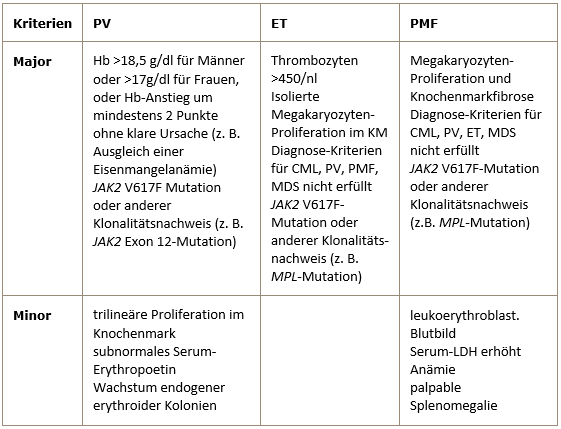
The diagnosis of PV requires either both major criteria and one minor criterion or the first major criterion and two minor criteria.
The diagnosis of ET requires all 4 major criteria.
The diagnosis of PMF requires all 3 major criteria and two minor criteria. -
The term myelodysplastic syndromes (MDS) covers a heterogeneous group of clonal myeloid diseases. A common feature of these diseases is the maturation disorders of myelopoiesis in the bone marrow, combined with peripheral cytopenia. MDS must be differentiated from acute myeloid leukemia (AML) on the one hand and from myeloproliferative neoplasms (MPN) on the other. The basic diagnostics for suspected MDS include
- Bone marrow cytology
- Cytogenetics of the bone marrow
- Microscopic differential blood count
- Bone marrow histology
- Immunophenotyping (flow cytometry) of the bone marrow
- Molecular genetics
The two most important bone marrow examinations in the diagnosis of MDS are morphology and cytogenetic analysis. Bone marrow histology should be analyzed by a hematopathologist.
Historically, MDS was also referred to as “preleukemia”. This term stems from the fact that all MDS carry the risk of developing into acute myeloid leukemia (AML). However, this risk varies greatly depending on the type of MDS. The transition from MDS to AML is fluid and the differential diagnosis of MDS<->AML must ultimately also be made clinically. If there are 20% or more blasts in the bone marrow, it is defined as AML; if there are less than 20%, it is MDS. The microscopic differential blood count is also part of the primary diagnosis. This reflects the type and number of cytopenias (important for MDS classification), and the number of monocytes must also be taken into account. A persistent monocytosis >1000/µl is defined as chronic myelomonocytic leukemia (CMML), which is counted among the MDS/MPN mixed forms.
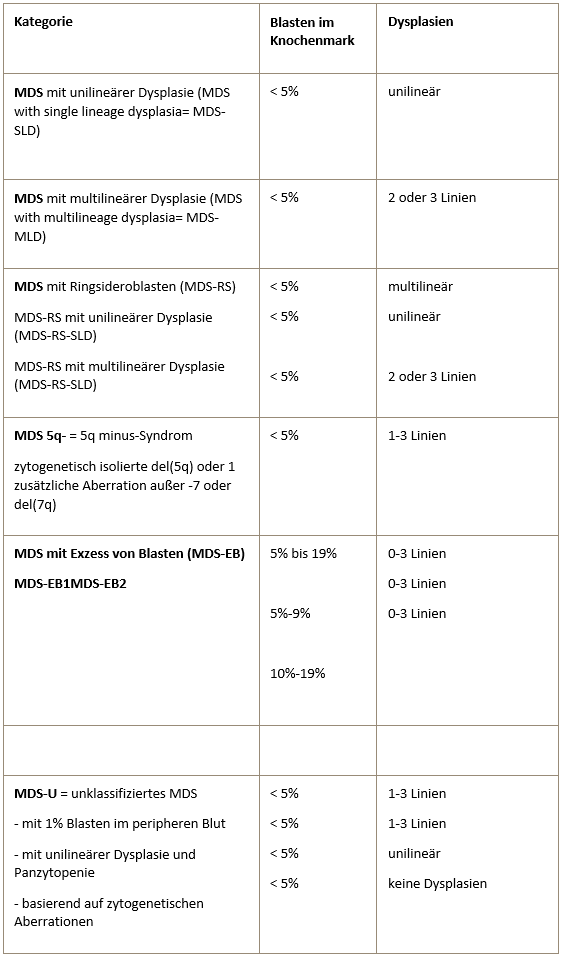
WHO classification (2016) of myelodysplastic syndromes
Bone marrow cytology
Bone marrow cytology is of great importance in the diagnosis and assessment of the course of MDS. On the one hand, the proportion of blasts and, on the other, the dysplastic changes in the various cell rows are assessed. It also detects the presence of ring sideroblasts. Ring sideroblasts are erythroblasts that show perinuclear granules (so-called siderosomes) in the Berlin blue stain. These granules are mitochondria loaded with ferritin. Small numbers of ring sideroblasts are also found in healthy bone marrow. In MDS, they are an expression of an iron utilization disorder.
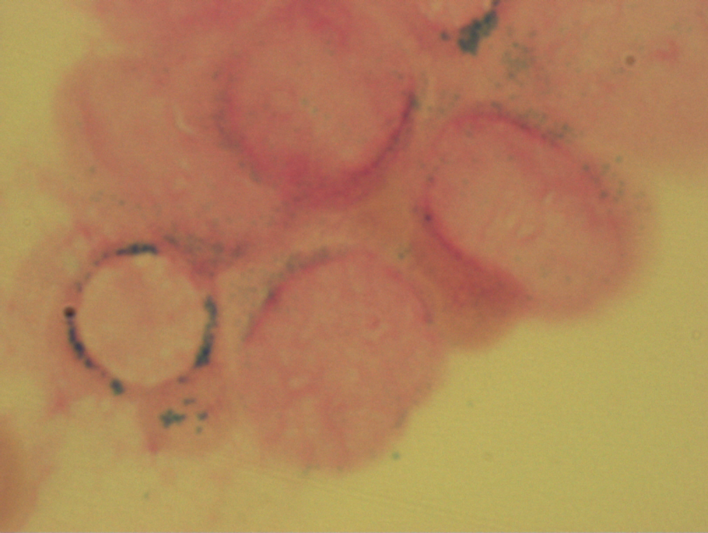
Ringsideroblasts with RARS
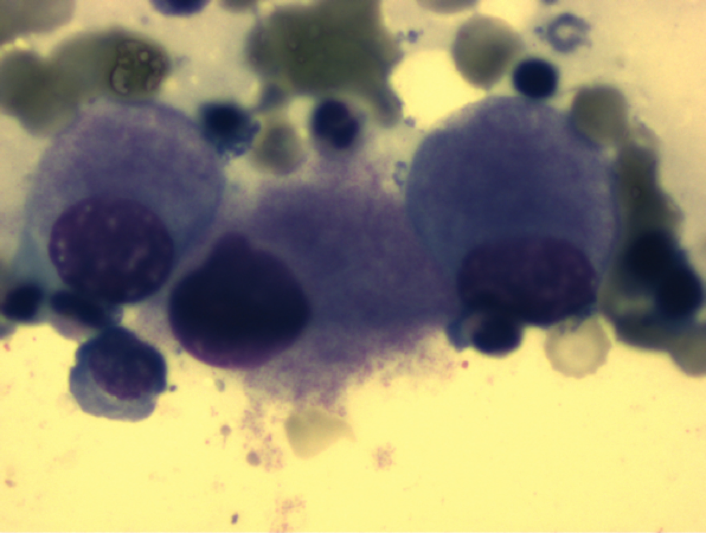
Maturation impaired megakaryocytes in MDS 5q-
Flow cytometry
Immunophenotyping by flow cytometry should be performed additionally, also to estimate the proportion of myeloid blasts.
Genetics
The genetic basis of myelodysplastic syndromes is heterogeneous. A large number of genetic changes are known, some of which also have prognostic significance and which also occur in variable combinations. Up to now, the cytogenetically defined changes have been particularly important, as clear risk groups could be identified here. Therefore, cytogenetic analysis in MDS is as important as bone marrow morphology. A thorough cytogenetic analysis should be carried out whenever MDS is suspected. Bone marrow cytogenetics should also include fluorescence in situ hybridization (FISH) for the most important cytogenetic aberrations occurring in MDS.
Prognostic value of common cytogenetic aberrations in MDS

All aberrations not included in this table are assigned to the intermediate prognostic risk group.
The panel of FISH probes for MDS diagnostics in the Berlin laboratory includes the following probes/gene loci:
- 5q31/5q33 (5q deletion, monosomy 5)
- 7q31 (7q31 deletion, monosomy 7)
- 20q12 (20q12 deletion, monosomy 20)
- CEP 8 (trisomy 8)
- TP53 (17p13 deletion)
- CEN Y (loss of the Y chromosome)
- TEL (ETV6) (12p13 deletion)
5q minus syndrome
The most common cytogenetic aberration in MDS are deletions of the long arm of chromosome 5 (del(5q)). A “minimally deleted region” was localized in the 5q31-32 area.
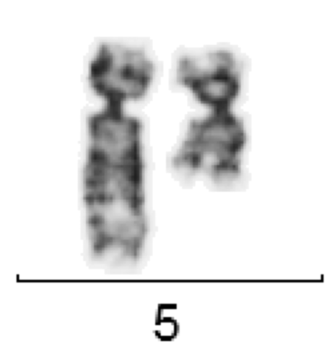
del(5)(q12Q33) for MDS
It is still largely unknown what role the genes located in this region play in the molecular pathogenesis of MDS. The 5q minus syndrome (MDS 5q) is characterized by a partial or complete del(5q) as the only cytogenetic aberration (i.e. without further additional aberrations) or with an additional aberration (except -7 or del(7q)).the proportion of blasts in the BM is less than 5%. In MDS 5q-, unusually small megakaryocytes with monolobulated nuclei are typically found in the bone marrow.
Molecular genetics
In recent years, many molecular genetic aberrations have been described in MDS. The affected genes include splicing factors such as SRSF2 or SF3B1, tumor suppressors such as TP53, epigenetic regulators such as TET2, DNMT3A or ASXL1, transcription factors such as RUNX1, and others. However, each of these gene aberrations is present in less than 10-20% of MDS cases. Relatively clear unfavorable prognostic implications are known for some genes (TP53, ASXL1, EZH2). However, most of these aberrations are not specific to MDS and are also found in other myeloid neoplasms (AML, MPN). AML-typical aberrations such as t(15;17)/PML-RARA, inv(16)/CBFB-MYH11, t(8;21)/AML1-ETO do not occur in true MDS. Molecular genetic examinations (typically as a “gene panel”) in MDS can be used to confirm the diagnosis, to estimate the prognosis, to obtain molecular markers for possible later quantitative observations of the course of the disease and also to identify target genes/proteins for possible targeted therapy.
-
MDS-MPN mixed molds
There are malignant hematological diseases that have characteristics of both myeloproliferation (visible increase in at least one cell row in the peripheral blood count) and myelodysplasia (dysplastic changes in myelopoiesis in the bone marrow). In the 2008 WHO classification, the entity “myelodysplastic/myeloproliferative neoplasms” was therefore created. This entity must be differentiated from MPN and MDS. Bone marrow diagnostics are the same as for MPN and MDS. Two somewhat more precisely defined forms of MDS/MPN are briefly described as examples.
Chronic myelomonocytic leukemia (CMML)
CMML is defined by the following characteristics:
- Long-term persistent monocytosis > 1000/µl in the peripheral blood without any other recognizable cause
- Dysplasia in one or more rows of hematopoiesis in the bone marrow
- No detection of BCR-ABL or a Philadelphia chromosome
- Proportion of blasts in the bone marrow < 20%
The genetic background of CMML is not uniform and has not yet been clearly deciphered. In a small minority of cases (less than 5 %), the JAK2 V617F mutation is detectable. Bone marrow cytology and cytogenetic examination are of particular diagnostic importance.
Refractory anemia with ring sideroblasts and thrombocytosis (RARS-T)
The RARS-T is defined by the following features:
- < 5 % Blasts in the bone marrow
- Dysplasia in one or more cell lines in the bone marrow
- > 15 % ring sideroblasts in the bone marrow
- Thrombocytosis > 600/nl in peripheral blood
The most important differential diagnoses are essential thrombocythemia (ET; no high-grade dysplasia and no proliferation of ring sideroblasts) and RARS or RCMD (these show no thrombocytosis).
In the majority of cases, the JAK2 V617F mutation can be detected in RARS-T.
-
Some malignant hematologic diseases are associated with eosinophilia. This is particularly characteristic of myeloproliferative neoplasms. However, eosinophilia is also found in some T-cell lymphomas, for example.
The basic diagnostics for suspected malignant disease with eosinophilia include
- Bone marrow histology
- Bone marrow cytology
- Immunophenotyping
- Molecular genetics
- Cytogenetics.
The genetic basis of malignant eosinophil diseases is heterogeneous and only partially known to date. In some cases, aberrations of certain genes are present:
- PDGFRA (platelet-derived growth factor receptor alpha, alpha receptor for platelet growth factor) on chromosome 4q12.
- PDGFRB (platelet-derived growth factor receptor beta, beta receptor for platelet-derived growth factor) on chromosome 5q33.1.
- FGFR1 (fibroblast growth factor receptor 1) on chromosome 8p12.
All 3 genes are tyrosine kinases. Presumably, other as yet unidentified tyrosine kinases are also involved in the etiopathogenesis of malignant eosinophil diseases.
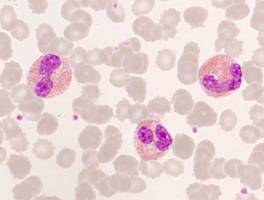
Eosinophils in peripheral blood
A precise cytogenetic analysis (preferably always from bone marrow) is of crucial importance. This cytogenetic analysis should include fluorescence in situ hybridization (FISH) for the PDGFRA, PDGFRB and FGFR1 genes.
A fusion gene FIP1L1-PDGFRA is found in a small proportion of cases of chronic eosinophilic leukemia. This fusion gene can be detected by molecular genetics using RT-PCR or FISH analysis. It is caused by a microdeletion on chromosome 4q12, which is not visible by conventional cytogenetics and in which the neighboring genes FIP1L1 and PDGFRA are fused together.
The detection of PDGFRA or PDGFRB aberrations has direct therapeutic consequences: both tyrosine kinases can be inhibited very well with imatinib, nilotinib or dasatinib.
Mast cell diseases
In the 2017 WHO classification of hematologic neoplasms, mastocytosis was defined as a separate disease entity (previously it had been classified under myeloproliferative neoplasms). A distinction is made between the following forms:
- Cutaneous mastocytosis
- Indolent systemic mastocytosis
- Systemic mastocytosis with associated hematologic neoplasia (SM-AHN)
- Aggressive systemic mastocytosis (ASM)
- Mast cell leukemia
- Mast cell sarcoma
In the vast majority of all forms of mastocytosis, activating somatic mutations are found in the tyrosine kinase KIT. By far the most common is the KIT D816V mutation, which can be detected with high sensitivity by quantitative PCR in blood, even in clinical forms that do not appear to show hematogenous seeding. If the KIT D816V mutation cannot be detected, but there is still a high degree of suspicion of a clonal mast cell disease, KIT sequencing should be performed, which can also detect other, rarer KIT mutations. Depending on the type of KIT mutation, various tyrosine kinase inhibitors can be used.
-
Bone marrow diagnostics for AML includes
- Bone marrow cytology
- Immunophenotyping
- Molecular genetics
- Cytogenetics.
Bone marrow histology is generally dispensable and only necessary in special cases (punctio sicca).
Cytomorphology
Cytomorphology is more important in AML than in ALL, as some subtypes of AML can already be diagnosed cytomorphologically. AML is often still classified according to the FAB (French-American-British) classification. This classification dates back to 1976 and is a purely cytomorphological classification. More modern classifications, such as the WHO classification from 2016, also take genetic and immunological aspects into account. Cytomorphology is particularly important in the detection of the FAB-M3 subtype (acute promyelocytic leukemia). The FAB-M4Eo subtype (acute myelomonocytic leukemia with abnormal eosinophils) can also usually be recognized by cytomorphology.
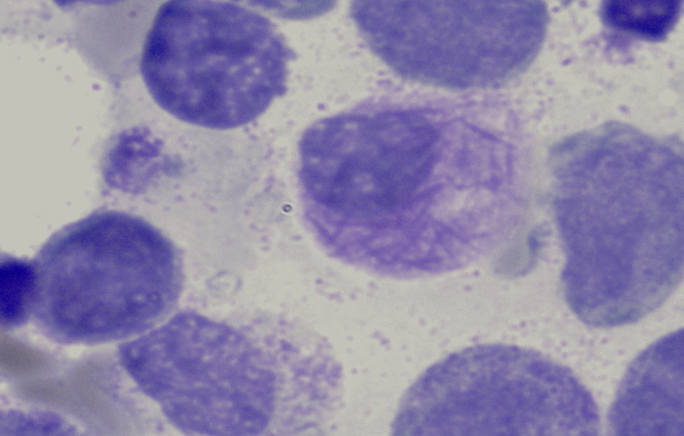
Bundle of Auer rods for AML M3
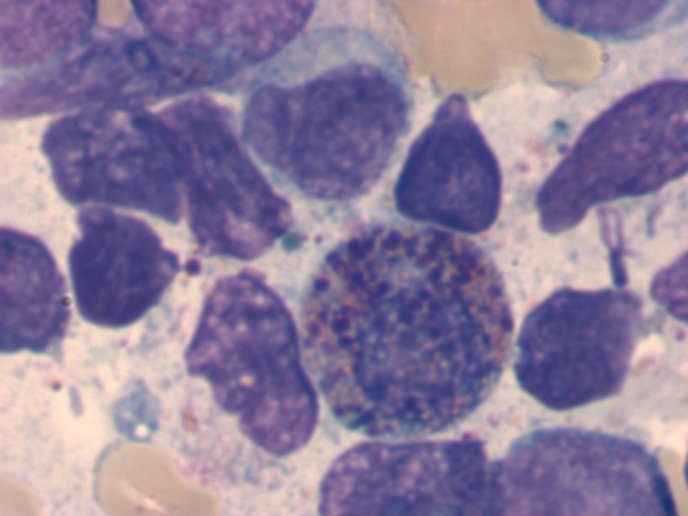
Abnormal eosinophil in AML M4Eo
Flow cytometry
Immunophenotyping using flow cytometry can confirm that an acute leukemia belongs to the myeloid series. However, flow cytometry is less important in the further subtyping of AML in routine diagnostics than in ALL, as the individual AML subtypes (FAB or WHO) cannot be differentiated from each other with sufficient certainty by immunocytology.
Cytogenetics and molecular genetics
Cytogenetics and molecular genetics in particular are becoming increasingly important for AML. A large number of genetic aberrations have now been described. This makes the picture even more confusing because these genetic aberrations can often occur in combination.
The basic genetic diagnosis for every AML should always include a conventional cytogenetic analysis. The following table lists the indications for molecular genetic testing.
Indications for molecular genetic testing
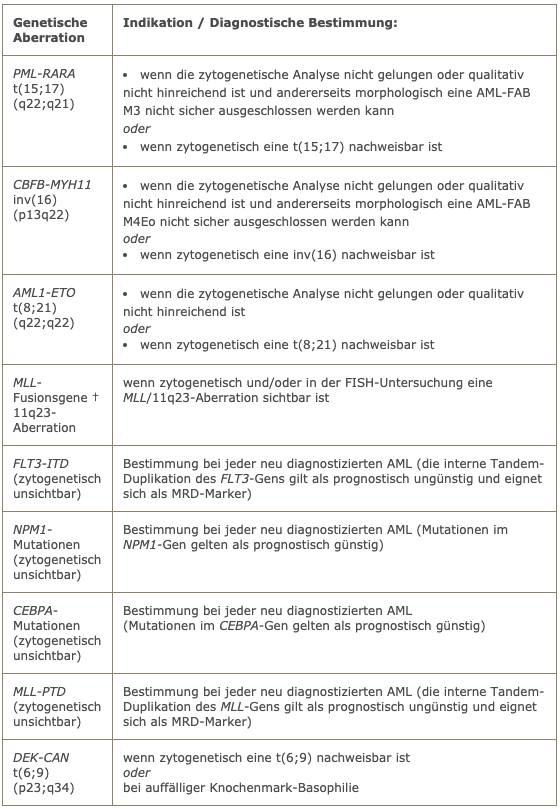

† are the most common MLL fusion genes in AML:
- MLL-AF9/t(9;11)(p22;q23)
- MLL-ENL/t(11;19)(q23;p13.3)
- MLL-ELL/t(11;19(q23;p13.1)
- MLL-AF6/t(6;11)(q27;q23)
- MLL-AF10/t(10;11)(p13;q23)
Together they account for more than 80% of all MLL translocations in AML.
-
Bone marrow diagnostics for ALL includes:
- Bone marrow cytology
- Immunophenotyping
- Molecular genetics
- Cytogenetics.
Bone marrow histology is generally dispensable and only necessary in special cases (punctio sicca).
Around a good third of all cases of ALL occur in childhood (< 15 years), the rest in adolescence and adulthood (≥ 15 years). Precise immunological and genetic characterization serves to confirm the diagnosis as well as to stratify the risk, as it has been shown that different ALL subtypes have a very different prognosis. In addition, genetic-immunological characterization opens up possibilities for targeted therapy (e.g. imatinib, rituximab, blinatumomab).
Flow cytometry
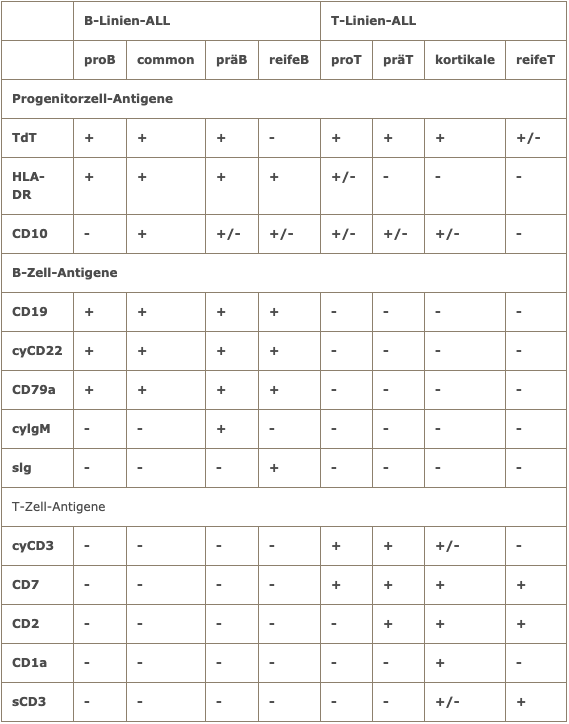
Flow cytometric analysis is of central importance in ALL. It allows the differentiation between B-line and T-line ALL. Within B-lineage ALL, the differentiation of mature B-ALL from B-precursor ALL is of great therapeutic relevance.
Immunological classification of ALL according to EGIL (European Group for the immunological classification of acute leukemias)
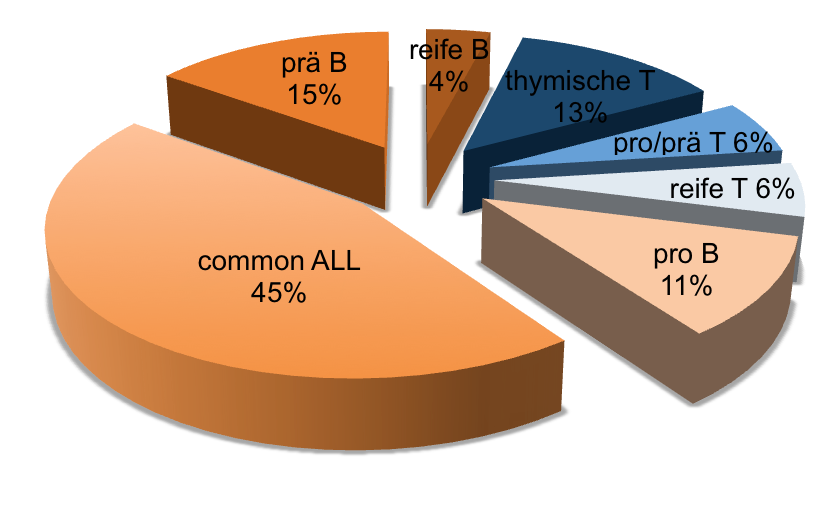
Approximate frequency of different immunophenotypes in adult ALL
The test material for flow cytometry is bone marrow aspirate or blast-rich peripheral blood. In special cases, (unfixed) lymph nodes can also be examined.
Genetics
The genetic background of ALL in childhood and in adulthood and adolescence differs considerably. Many genetic aberrations in ALL show a pronounced age dependency. Accordingly, the diagnostic focus is usually somewhat different for children and adults.
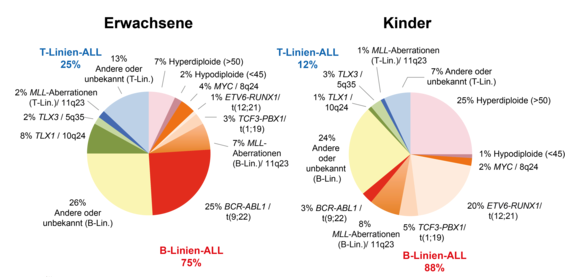
Approximate distribution of genetic aberrations in ALL
The genetic aberrations found in ALL are often closely correlated with certain immunophenotypes.
Frequent genetic aberrations in ALL
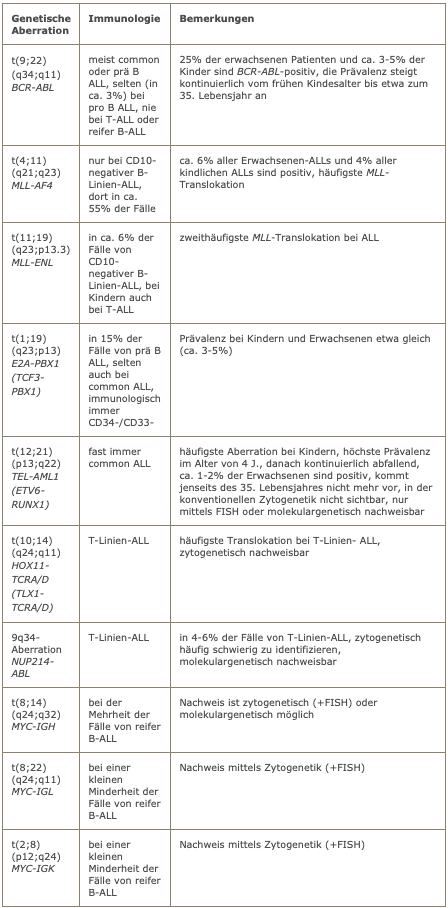
Cytogenetics
The cytogenetic examination is obligatory. In the case of B-precursor ALL, it should also include a FISH test for BCR-ABL and a FISH test for MLL aberration. A FISH test for MYC translocation should be performed in mature B-ALL.
Molecular genetics
The molecular genetic testing program is based on the immunophenotype. In the case of B-precursor ALL, testing for BCR-ABL is mandatory. In CD10-negative ALL, the two most common MLL fusion genes, i.e. MLL-AF4 and MLL-ENL, should be examined. Pre-B-ALL should be tested for E2A-PBX1. In mature B-ALL, the MYC-IGH fusion should be investigated. In the case of T-line ALL, testing for NUP214-ABL is recommended.
-
Non-Hodgkin’s lymphomas form a heterogeneous group of diseases. Basic diagnostics include:
- Lymph node and/or bone marrow histology
- Immunophenotyping (flow cytometry)
- Cytogenetics
- Molecular genetics
- Bone marrow cytology
Lymph node or bone marrow histology, which is analyzed by a pathologist, is usually of particular importance. In difficult cases, a reference pathologist for lymphoma diseases should always be consulted. Bone marrow diagnostics are otherwise used to confirm the diagnosis on the one hand and to diagnose the spread of the disease (staging) on the other.
In some leukemic lymphomas, the diagnosis can be made from the blood. In any case, a cytogenetic analysis should be performed if vital tumor material is available.
Immunophenotyping using flow cytometry
Peripheral blood, bone marrow, pleural effusion or ascites are suitable starting materials for flow cytometric immunophenotyping if they show evidence of lymphoma. In principle, this is also possible without any problems using unfixed lymph node tissue, but is usually not used in practice, as extirpated lymph node tissue is usually fixed immediately, after which immunohistochemical staining is performed by the pathologist. In leukemic lymphomas (e.g. chronic lymphocytic leukemia, Waldenström’s disease, Sézary syndrome, leukemic forms of: Immunocytoma, follicular lymphoma, mantle cell lymphoma) or aleukemic lymphomas with regular bone marrow involvement (e.g. multiple myeloma), immunophenotyping can be performed from blood or bone marrow. Immunophenotyping can be used to differentiate between T-series lymphomas and T-cell or NK-cell lymphomas. In addition, a certain allocation to the degree of maturity and differentiation is possible. In some cases, the diagnosis can even be made by flow cytometry (hairy cell leukemia). Most lymphomas have a relatively characteristic immune marker profile. However, there are also atypical antigen patterns in a considerable number of cases.
Cytogenetics and molecular genetics
While purely histopathological, i.e. morphological, characterization used to be at the forefront of lymphoma diagnostics (later supplemented by immunohistochemistry and immunophenotyping), genetic examinations have become increasingly important in recent years. Genetic aberrations in NHLs are sometimes relevant in terms of prognosis or differential diagnosis and thus influence treatment decisions. A comprehensive lymphoma diagnosis should therefore include a genetic analysis, particularly at the time of initial diagnosis.
The cytogenetic examination requires vital, divisible tumor tissue. This can be unfixed lymph node tissue or bone marrow aspirate, for example. Molecular genetic diagnostics for NHL is often (but not necessarily) based on the findings of cytogenetic diagnostics. The test material must also be unfixed for molecular genetic diagnostics.
Common cytogenetic and molecular aberrations in non-Hodgkin’s lymphomas
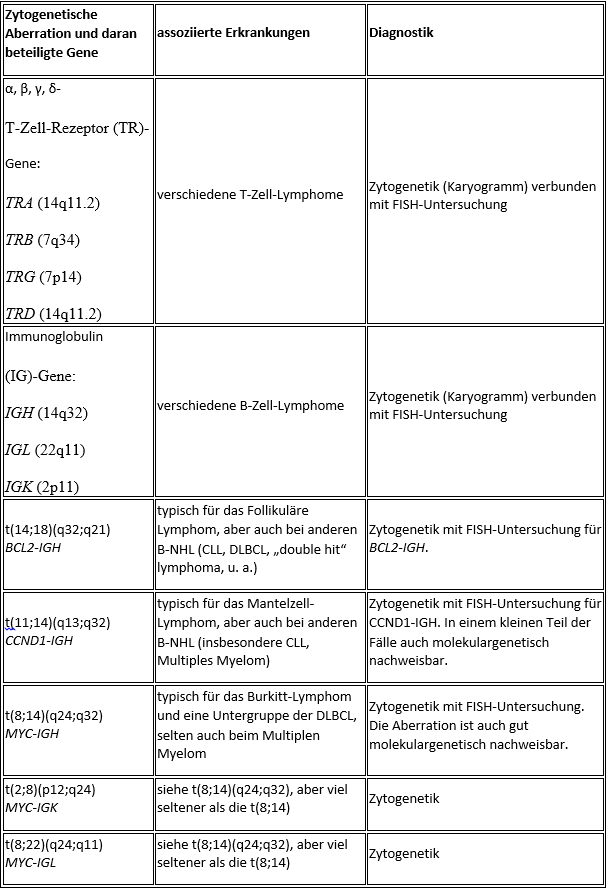
Individual selected lymphoma entities
Chronic lymphocytic leukemia (CLL)
The diagnosis of CLL can usually be made by flow cytometry based on the typical immunophenotype (CD5+/CD10-/CD19+/CD23+). Genetic testing is particularly useful for younger patients, as it allows individual high-risk patients to be identified. TP53 aberrations, which can appear cytogenetically as a deletion (del(17p13)), are primarily considered a high-risk feature. The loss of the long arm of chromosome 11 (11q-) is also considered to have an unfavorable prognosis, while del(13q) or trisomy 12 as isolated aberrations indicate a rather favorable prognosis.Follicular lymphoma (FL)
Follicular lymphoma is classified into 3 grades according to the proportion of blasts and growth pattern (follicular versus diffuse). A grade 3 FL corresponds to a highly malignant NHL. Genetically, the translocation t(14;18) with BCL2-IGH fusion is detectable in the vast majority of cases. Detection is possible by cytogenetics (+ FISH) and in the majority of cases also by molecular genetics using PCR. If the translocation can also be detected by molecular genetics, this molecular marker can be used to carry out follow-up examinations for minimal residual disease (MRD). Flow cytometry usually shows a CD5-/CD10+/CD19+/CD20+/CD23-/sIg+ immunophenotype. Histopathologically, BCL2 and BCL6 are typically positive.Mantle cell lymphoma (MZL)
By flow cytometry, mantle cell lymphoma shows a CD5+/CD10-/CD19+/CD23- immunophenotype. Immunohistochemically, cyclin D1 (CCND1) is typically positive.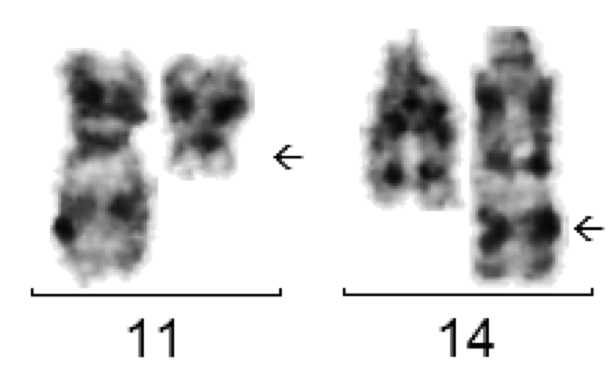
The translocation t(11;14) is typically found in mantle cell lymphoma but also in other NHLs.
Genetically, more than 90% of cases have the translocation t(11;14) with CCND1-IGH fusion. Cytogenetic detection is easy to perform. The fusion can only be detected molecularly by PCR in one of the cases, as the chromosome breakpoints are widely dispersed. Nevertheless, a molecular genetic examination should be performed if t(11;14) is cytogenetically detectable, as this may allow the identification of a molecular marker for quantitative follow-up examinations.
Burkitt’s lymphoma (BL)
The differential diagnosis between Burkitt’s lymphoma and diffuse large B-cell NHL (DLBCL) is sometimes difficult. Great importance is attached to morphology and genetic analysis. Cytogenetically, there are almost always translocations of the MYC gene on the long arm of chromosome 8 (8q24) near the immunoglobulin genes IGH (heavy chain locus, 14q32), IGK (light chain locus kappa, 2p12) or IGL (light chain locus lambda, 22q11).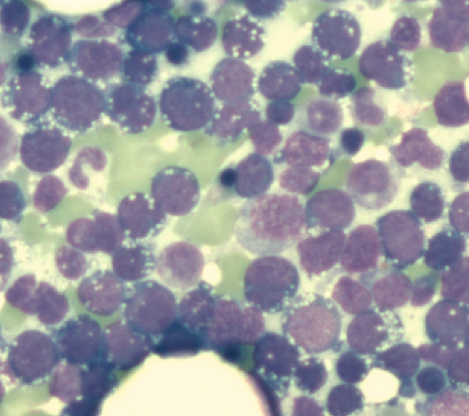
Bone marrow infiltration due to Burkitt’s lymphoma. The blasts show numerous cytoplasmic vacuoles and highly basophilic cytoplasm.
In about 75-85% of BL cases, a translocation t(8;14)(q24;q32) is present, which can be detected both cytogenetically and molecularly. Cytogenetically, a translocation t(2;8)(p12;q24) is found in about 5% of cases and a translocation t(8;22)(q24;q11) in about 10%. However, all three translocations also occur in other NHL entities (DLBCL, rarely multiple myeloma) and therefore do not prove BL on their own. Cytologically, BL blasts are usually impressive in the form of a “FAB L3 morphology”.
Diffuse large B-cell lymphoma (DLBCL)
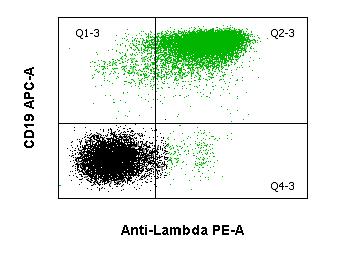
Diffuse large B-cell lymphoma is an umbrella term for a group of highly malignant B-cell lymphomas with a diffuse growth pattern. DLBCLs form a biologically heterogeneous group. Morphologically, a distinction is essentially made between the centroblastic, immunoblastic and anaplastic variants. B cell antigens (CD19, CD20, CD79a) can be detected by flow cytometry, as well as surface immunoglobulin (sIg), possibly with light chain restriction. CD10 is expressed in a variable percentage. Genetically, the most frequently affected loci are 3q27 (BCL6), 18q21 (BCL2) and 8q24 (MYC) and the immunoglobulin heavy chain locus (IGH).Hairy cell leukemia (HCL)
Hairy cell leukemia is a rare low-malignant B-NHL. In the bone marrow examination, often only bone marrow blood can be aspirated and morphologically typical hair cells are not always found in the peripheral blood.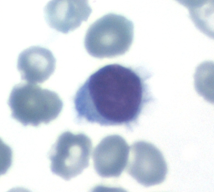
Hair cell in peripheral blood
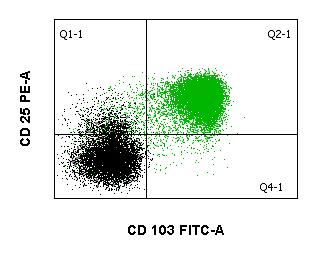
The diagnosis is usually made immunologically, e.g. by flow cytometry, in conjunction with typical clinical findings, as HCL is characterized by the relatively specific expression of the CD103 antigen. The antigens CD19, CD20, CD22, CD11c, CD25, and FMC7 are also expressed, while CD5, CD10 and CD23 are typically absent. Genetically, the BRAF V600E mutation, which is also found in a number of solid tumors, is usually detectable in typical HCL. If HCL is suspected, molecular genetic testing for BRAF V600E mutation should be performed in addition to flow cytometry.
Multiple myeloma (MM)
Multiple myeloma is a clinically and diagnostically multifaceted disease. Plasma cell antigens (e.g. CD38 and CD 138) can be detected by flow cytometry.Typically, a CD19-/CD79a+/CD38+/CD138+ immunophenotype is present. Aberrant antigen expressions (CD117, CD20, CD52, CD10) are occasionally observed. The disease can usually be clearly diagnosed by bone marrow morphology.
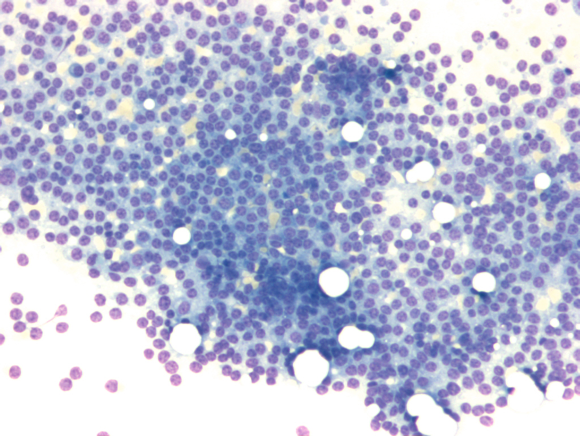
High-grade bone marrow infiltration due to multiple myeloma
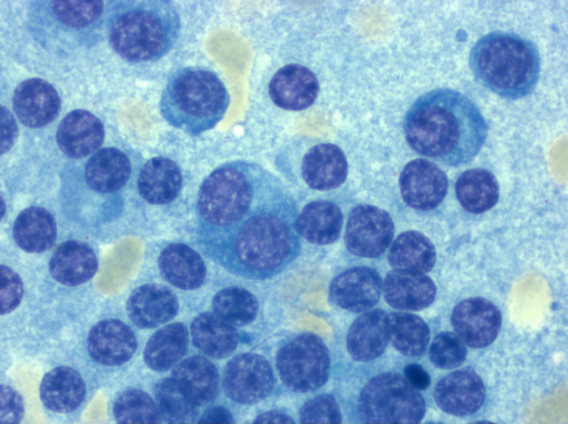
Detail enlargement
For a long time, the genetic analysis of MM was hampered by the fact that myeloma cells in culture divide very slowly or not at all, so that cytogenetic analysis was often not possible.
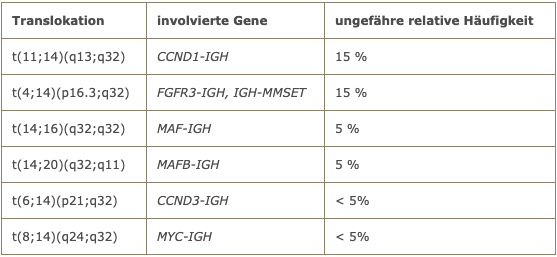
The Berlin laboratory is using a special culture medium with which a significantly improved rate of evaluable metaphases can be achieved with myeloma cells in culture. In slightly more than half of the cases, chromosome translocations involving the immunoglobulin heavy chain locus (IGH) are present. The most common chromosome translocations in multiple myeloma are listed in the following table.
Chromosome translocations in multiple myeloma
Prolymphocytic leukemia (PLL)
Prolymphocytic leukemia can have a B-cell or a T-cell immunophenotype. Morphologically, the B-PLL largely shows immature medium-sized prolymphocytes with prominent nucleoli. Immunophenotypically, B-cell antigens are expressed (CD19/CD22/CD79a+b/FMC7) and light chain restriction is detectable. In contrast to CLL, CD5 and CD23 are only detectable in a minority of cases. Genetically, complex karyotypes are often found. T-PLL has a CD2+/CD3+/CD7+/CD52+ phenotype. CD4+/CD8- expression is usually found, but occasionally CD4 and CD8 are co-expressed. Cytogenetic aberrations of the T-cell receptor genes are typical.Sézary syndrome
A Sézary syndrome, i.e. a leukemized cutaneous T-cell lymphoma, is present if morphologically typical Sézary cells are detectable in the peripheral blood and a neoplastic T-cell population is detectable by flow cytometry. According to the 2008 WHO classification, the diagnosis requires at least 1000/µl Sézary cells and/or a CD4+/CD8+ ratio of at least 10:1 and/or the loss of several T-cell antigens (e.g. CD7). Even with Sézary cells circulating in the blood, there is usually no evidence of bone marrow infiltration by the underlying T-cell lymphoma. Typically, a CD2+/CD3+/CD5+/CD7-/CD4+ immunophenotype is present. CD8+ positive cases are rare. Morphologically, in contrast to normal lymphocytes, Sézary cells show strongly changing, sometimes bizarre nuclear shapes. Genetically, Sézary syndrome appears to be heterogeneous and has so far been poorly characterized. -
PNH is caused by an acquired mutation in the PIGA gene on Xp22.1. This results in a disruption of the anchoring of glycoproteins in the cell membrane (the so-called GPI anchor is disrupted). The proteins affected include CD55 and CD59, which have a complement-inhibiting function. As a result of the lack of CD55 and CD59, PNH patients experience chronic or seizure-like complement activation. The main symptoms of PNH are Coombs-negative haemolysis, a tendency to thrombosis (often with atypical localization), peripheral cytopenia of one or more rows and possibly also renal dysfunction (due to microthrombi). The symptoms of PNH patients depend crucially on the size of the PNH clone. It is not uncommon for PNH to occur in the context of other hematological diseases – often with aplastic anemia, less frequently or to a lesser extent with low-risk myelodysplastic syndromes.
Flow cytometry
The diagnostic “gold standard” for the diagnosis of PNH is the flow cytometric examination of peripheral blood for the expression of GPI-anchored proteins. The DGHO guidelines (as of March 2012) require the examination of at least 2 different markers (GPI-anchored proteins or GPI anchors themselves). At least granulocytes and erythrocytes should be examined. The proportion of the PNH clone can best be estimated from the proportion of GPI-deficient granulocytes, as this is not influenced by hemolysis or transfusions.
Sample logistics are important: the sample sent in must be as fresh as possible, i.e. analyzed within 48 hours. Sampling on Fridays or before public holidays should therefore be avoided. For transportation times > 24 hours, the sample should be cooled (+1 to +10 oC).
Indications for diagnostics
- Coombs-negative hemolysis
- Thrombosis if at least one of the following criteria is met:
- Atypical localization (sinus vein thrombosis, Budd-Chiari, etc.)
- Thromboses in connection with unclear cytopenia
- Thromboses of unknown cause, arterial thromboses
- Suspicion of aplastic anemia
- Suspected myelodysplastic syndrome
- Recurrent pain with concomitant signs of hemolysis




contact
-
Labor Berlin – Charité Vivantes GmbH
Sylter Straße 2
13353 Berlinphone: +49 (30) 40 50 26-800
fax: +49 (30) 40 50 26-615 -
Campus Charité Mitte (CCM)
phone: +49 (30) 450-513023
fax: +49 (30) 450-513933 -
Campus Virchow Clinic (CVK)
Morphology
phone: +49 (30) 40 50 26-498
fax: +49 (30) 40 50 26-615Immunophenotyping
phone: +49 (30) 40 50 26-497
fax: +49 (30) 40 50 26-615Tumor genetics
phone: +49 (30) 40 50 26-493
fax: +49 (30) 40 50 26-618Tumor cytogenetics
phone: +49 (30) 450-569145
fax: +49 (30) 450-569996
Pre-analytical notes and downloads
-
The anticoagulant for all cytological examinations is EDTA. Citrate is also possible in exceptional cases. Heparin, on the other hand, is unfavorable and should not be used.
Suitable test materials are
- Bone marrow aspirate (5 ml)
- Peripheral blood (routine EDTA blood tubes)
- Effusion material (ascites, pleural effusion, pericardial effusion, etc.)
- Liquor
Care should be taken to ensure that the transportation time does not exceed 24 hours and that the material arrives at the laboratory before weekends by Friday, 14:00 at the latest. In principle, material should be sent with an express service (overnight service) to avoid excessively long transportation times.
-
Flow cytometry
The antigoagulant for all flow cytometric tests is EDTA. Heparin is less suitable and should only be used if nothing else is available (e.g. accidental collection in the wrong syringe during a bone marrow puncture).
Suitable test materials are
- Bone marrow aspirate (5 – 10 ml)
- Peripheral blood (5 – 10 ml)
- Effusion material (ascites, pleural effusion, pericardial effusion, etc.), if possible 10 ml
- Liquor
PNH diagnostics are performed from peripheral blood.
Samples should always be shipped using an express service (overnight service) in order to keep transportation times as short as possible. The samples should not be older than 24 hours when they arrive at the laboratory. Before weekends, please ensure that the sample arrives at the laboratory by noon on Friday at the latest.
-
The anticoagulant for all molecular genetic tests is EDTA. Citrate can also be used in exceptional cases. Heparin, on the other hand, is unfavorable and should only be used if nothing else is available (e.g. accidental collection in the wrong syringe during a bone marrow puncture).
In principle, the following can be used as test material:
- Bone marrow aspirate (5 ml)
- Peripheral blood (10 ml)
- Effusion material (ascites, pleural effusion)
- Liquor
- Lymph node or tumor biopsy (unfixed !)
If tumor material or lymph nodes are sent in, please contact us in advance by telephone. Molecular genetic diagnostics from cerebrospinal fluid usually only has a chance of success if it is very rich in cells. Here too, prior consultation by telephone is recommended.
Samples should be sent by express mail, especially for time-critical samples (acute leukemia). If possible, shipping should not take place on a Friday.
Requisition slips and downloads
Services for private patients, self-paying patients or elective laboratory services will be invoiced by LABOR BERLIN directly to the respective payer, unless otherwise agreed with the sender. For this purpose, the sender shall forward the necessary patient data to LABOR BERLIN and ensure that the patients are informed about the possible forwarding of laboratory orders to LABOR BERLIN and the associated organizational measures, including billing by a private medical clearing office, in the manner prescribed by law and consent to this. The legal requirements with regard to the free choice of doctor are taken into account. We would like to point out that, in accordance with the provisions of the German Hospital Remuneration Act (KHEntgG), external elective laboratory services must be ordered by the sender on a case-by-case basis and specifically by the elective physicians concerned.
-
Haematology, Cytology & Flow Cytometry (CVK site)
pdf -
Specialized Hematological Diagnostics
pdf -
Consent slip for liquid biopsy
pdf -
Requisition Slip Hämatologie Zytologie (Standort CVK)
pdf -
Hematology requisition slip (CCM location)
pdf -
Declaration of consent for tumor cytogenetics and tumor genetics
pdf -
Requisition slip for hematology combination certificate Cytology & Molecular Genetics (CVK site)
pdf -
DPD diagnostics requisition slip
pdf