Human Genetics


Molecular genetics and tumour cytogenetics
The Department of Human Genetics emerged from the human genetics diagnostic laboratories of the Institute of Medical Genetics and the Institute of Human Genetics at Charité-Universitätsmedizin Berlin. The Molecular Genetics and Tumour Cytogenetics departments are also committed to Labor Berlin’s goal of offering cutting-edge diagnostics at the cutting edge of science and technology for our patients and senders. Molecular and tumour cytogenetic diagnostics play a key role in modern human medicine, which aims to meet the demand for individual, tailor-made treatment.
With a team of doctors and scientists, we offer a comprehensive range of diagnostic methods and human genetic expertise in the fields of molecular genetics and tumour cytogenetics. Doctors and consignors can obtain personalised and expert advice on all clinical and diagnostic issues from designated contact persons on our website.
For more information on the range of services / indications of Molecular Genetics, please visit the German version of this page
Unclear diagnosis
-
Contact person:
- Dr. rer. nat Jörg Schuldes (Joerg.Schuldes@laborberlin.com)
- Dr. rer. medic. Johannes Grünhagen (johannes.gruenhagen@laborberlin.com)
Analysing all coding sections of the genome (Whole Exome Sequencing) allows the data to be used flexibly for a wide range of indications. Complex issues such as unexplained developmental disorders, polygenic disease patterns and particularly rare diseases benefit particularly from WES. WES offers the possibility of updating the scope of the analysed genes even years after a diagnosis has been made, if further genes become known as the cause of the original indication on the basis of new scientific findings, or to expand the scope if the original indication changes due to other or further clinical symptoms. Both expanding and updating a virtual panel based on a WES dataset can only be done after consultation.
Due to this fact, WES can be used flexibly for patients for whose disease no panel analysis is available. A precise definition of clinical information about the index patient in the format of HPO terms (HPO: Human Phenotype Ontology; hpo.jax.org) is required for the correct selection of disease-relevant genes (virtual gene panel). Based on the generated exome dataset, the bioinformatic narrowing down of the 25kb basic diagnostics allows the compilation of specific genes that are associated with the disease pattern according to the latest scientific findings. This individual panel can then be analysed regularly.
-
Patient information on additional findings in WES
pdf
Range of molecular diagnostics methods
-
Molecular genetic diagnostics is part of human genetics, the aim of which is to identify the molecular causes of genetic diseases in humans. Specific DNA analyses of certain genes can be used to identify changes (mutations) in patients that are responsible for the occurrence of a disease. The knowledge gained through molecular genetic diagnostics is extremely important for further genetic counselling of the family and subsequent e.g. predictive and/or prenatal diagnostics. To date, around 14,000 genes with suspected disease associations and around 6,000 diseases with a clearly defined molecular genetic cause are known (OMIM database).
The majority are very rare monogenic diseases that occur less frequently than 1 case per 10,000 people in the population. However, such rare diseases affect around 1% of all newborns worldwide.
Mutations underlying rare genetic diseases are often not yet described in databases and are sometimes difficult to assess. The longer it takes to diagnose these genes, the more the diagnosis benefits from the experience of the evaluators and the growing number of database entries. This facilitates the task of distinguishing disease-causing mutations from variants without disease significance. The methods used in mutation searches have developed rapidly over the past 10 years, primarily due to access to population data, the use of evaluation standards (ACMG criteria) and prediction algorithms (mutation taster, polyphen, DANN, FATHMM, various meta-scoring systems).In collaboration with medical colleagues and genetic counselling, Labor Berlin offers affected individuals and families diagnostics at the highest level. To enable diagnostics for the benefit of those affected, Human Genetics regularly and successfully participates in round robin tests, monitors progress and findings in this rapidly developing field and utilises the latest sequencing technologies on the market.
Various methods are used for molecular genetic analyses:
- Next Generation Sequencing
- Whole exome sequencing (WES)
- PCR (Polymerase Chain Reaction)
- Sequencing according to Sanger
- MLPA (Multiplex Ligation-dependent Probe Amplification)
- Quantitative Real-Time PCR
- Fragment analysis
-
NextGeneration Sequencing (NGS) is a new type of nucleic acid analysis technology that refers to high-throughput or parallel sequencing technology. In contrast to Sanger sequencing, several hundred million gene fragments from a wide range of patients can be sequenced simultaneously in one process. The process consists of two steps: In the first step, the DNA of relevant genes is enriched. The second step is the sequencing of the enriched DNA using NGS.
In order to enrich the coding genomic regions of interest, in-solution capture methods have become established. A mixture of artificially synthesised locus-specific oligonucleotides (so-called probes) is added in solution to the fragmented genomic DNA sample and hybridises selectively to the genomic regions of interest.
The probes are labelled in such a way that they, including the bound DNA fragments, can be physically bound and washed in a next step to remove unhybridised and excess DNA fragments. In the final step, the probes are removed and the genomic fragments can be sequenced.For sequencing, we use sequencing devices from the market leader Illumina. With the help of a high-performance IT infrastructure optimised for NGS, the raw data is then processed in our in-house bioinformatics GATK-based pipeline and evaluated using special analysis tools.
Genetic diagnostics benefits greatly from NGS, as the enrichment of genomic target areas increases diagnostic sensitivity, the parallel analysis of genes in so-called “gene panels” saves time and allows many samples to be processed more cost-effectively. NGS has therefore established itself as a standard sequencing technology that we use in human genetics for a wide range of diagnostic questions (see Diagnostic panels).
-
-
Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all coding regions (“exons”) in a genome (“exome”). The procedure consists of two steps: In the first step, the exonic DNA is enriched. The human exonic DNA consists of ~180,000 exons, which make up about 1-2% of the human genome (50 million base pairs). The second step is the sequencing of the exonic DNA using NGS (description in section Next Generation Sequencing (NGS))
In order to enrich the coding genomic regions of interest, in-solution capture methods have become established. A pool of artificially synthesised locus-specific oligonucleotides (so-called probes) is added in solution to the fragmented genomic DNA sample and hybridises selectively to the genomic regions of interest.
The probes are labelled in such a way that they, including the bound DNA fragments, can be physically bound and washed in a next step in order to remove non-hybridised and excess DNA fragments. In the final step, the probes are removed and the genomic fragments can be sequenced.
Whole exome sequencing diagnostics is a particularly useful method for patients with complex issues or unspecific symptoms, as there is often no suitable gene panel available to clarify the genetic cause. After years of unsuccessful diagnostics, whole exome sequencing can be the method of choice to identify the genetic cause of the disease.
-
Application in diagnostics: part of the NGS method, part of Sanger sequencing, part of MLPA
PCR (polymerase chain reaction) is a method used to amplify parts of the genetic material DNA in vitro, i.e. outside a biological system. PCR is used to amplify one or more short, precisely defined parts of a starting DNA. The section to be amplified can be a gene or the partial section of a gene or even non-coding DNA sequences. For PCR, the enzyme DNA polymerase is used, which is also essential in living cells for DNA amplification, i.e. the copying of genetic information.
PCR requires several components.
- The original DNA as a template
- Two or more primers
- A heat-resistant DNA polymerase
- Deoxyribonucleoside triphosphates, the basic building blocks of DNA
- MG2+ ions, without which the function of the polymerase is not guaranteed
- A buffer solution that ensures the most physiological reaction environment possible for the polymerase.
The term polymerase chain reaction now describes the process of a repetitive programme for the duplication of DNA. This cyclic programme is based on a thermal destabilisation of the DNA double strand at >95°C and a subsequent doubling of the DNA strand at approx. 60°C. A chain reaction takes place in this context. A chain reaction takes place in this context due to the fact that the products of one cycle represent the starting materials (templates) for the next cycle and thus exponential amplification can take place.
PCR is used as a basic method in Sanger sequencing, MLPA, NGS, quantitative PCR, fragment analysis and the methylation test.
-
Sanger sequencing or the Sanger dideoxy method is a method of DNA sequencing named after Frederick Sanger, an English biochemist. It is based on the principle of the chain termination method and allows the determination of the base sequence in a specific DNA molecule. It is not possible to analyse two different DNA sections in one process.
The principle of DNA sequencing according to Sanger consists of synthesising new DNA strands of different lengths, which can then be separated by size in a capillary sequencer to determine the sequence information. In order to generate DNA strands of different lengths, fluorescence-labelled stop nucleotides (dideoxy nucleotides) are used in addition to the nucleotide mix. For each of the four possible nucleotides adenine, guanine, cytosine and thymine there is an equivalent, specifically fluorescent dye-labelled stop nucleotide. The correct mixture of nucleotide mix and stop nucleotides theoretically produces any conceivably long sequence fragment in a PCR.
Due to the different fluorescence-labelled fragments depending on the nucleotide, the base sequence can be evaluated with the help of special software after size separation.
We use Sanger sequencing routinely to detect known familial mutations and to confirm questionable NGS variants.
-
With the exception of the sex chromosomes, the human diploid genome has chromosomes that occur in pairs. Normally, the same genes are present once on each of these pairs of chromosomes; this is also referred to as two alleles (or bi-allelic). However, processes during meiosis or mitosis can lead to unequal distributions of the DNA segments and thus to a loss or gain of DNA material on the chromosomes. This is also referred to as gene dosage differences compared to the normal state, the wild type. These gene dose differences cannot normally be detected using the conventional PCR method, as the chain reaction amplifies exponentially and initial dose differences become blurred towards the end of the amplification phase.
With the MLPA method, dose differences of up to 50 different nucleic acid fragments can be detected in one batch. It was first described in 2002 (Schouten et al., Nucl Acids Res 30:12e57) and is based on the ligation of two locus-specific probes on the DNA, which are joined to form a fragment in a dose-dependent manner.
The amount of sequence-specific hybridised and ligated probes is thus proportional to the copy number of the target sequence.
This is followed by PCR and capillary electrophoretic separation of the products according to size. The fragments can be assigned to a specific locus by their defined different sizes.
Dose differences can be calculated by reducing or increasing the peak heights and areas compared to the reference samples.
The range of MLPA kits offered by the manufacturer MRC-Holland is now very extensive. MLPA kits are commercially available for the detection of deletions and duplications of various genomic regions in the context of a wide range of clinical questions. For example, methylation-specific MLPAs (MS-MLPA), which can be used to determine the copy number as well as to analyse the methylation pattern (detection of imprinting defects) and to analyse tumours, represent an extension of the classic MLPA.
An alternative to determining the gene dose is Quantitative Real-Time PCR (see below).
-
-
Quantitative real-time PCR is an amplification method for nucleic acids that is based on the principle of the conventional polymerase chain reaction (PCR). However, it is additionally coupled with a fluorescence measurement in the exponential phase of the PCR and thus enables the quantification of gene segments or transcripts. We use a device from Life Technologies for our diagnostics. In this device, the detection of PCR products is either sequence-unspecific (e.g. via SYBR Green) or sequence-specific using fluorescence-labelled probes.
The increase in the fluorescence signal during the course of the reaction is proportional to the amount of PCR product formed and can be monitored in real time during the course of the reaction.
The dose of the starting material can therefore be measured in absolute terms or relative to an internal control sequence and external control sample. The advantage over commercially available kits for dose determination lies in the flexibility. Sequence-specific primers can be produced for any loci in the genome.
One disadvantage of this method is its low specificity, as it is not possible to differentiate between desired and undesired PCR products. Parallel measurements of several loci (multiplex measurements) cannot be carried out either. We compensate for the first disadvantage by carrying out a melting curve analysis, which can be used to determine the fragment length(s) and thus the specificity.
An alternative to determining the gene dose is MLPA (see above).
-
Fragment analysis is based on the functional principle of the polymerase chain reaction (PCR). In contrast to classical PCR, sequence-specific fluorescence-labelled primer pairs are used for fragment analysis. By adding a fluorescence-labelled length standard and separating these labelled PCR products according to size in a capillary sequencer, it is possible to distinguish the size of just one base, in contrast to gel electrophoresis. By using several different fluorescent dyes, three-digit target sequences can be analysed in parallel in one PCR approach.
Fragment analysis is used in chimerism analysis, monosomy diagnostics, chromosome aberration tests, paternity tests and parentage analyses.



TUMOUR CYTOGENETICS
Range of services / Indication
-
Acute myeloid leukaemia (AML)
Acute myeloid leukaemia is a heterogeneous disease that can develop de novo, be therapy-associated (t-AML) or arise secondarily from a pre-existing myeloproliferative disease or MDS (s-AML). The incidence increases with age and is around 3.7 cases per 100,000 inhabitants per year, with a median age of 72 years.
According to the WHO classification 2017, AML is divided into the following subgroups
Acute myeloid leukaemia (AML) and related neoplasms
AML with recurrent genetic aberrations
- AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1
- AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11
- Acute promyelocytic leukaemia (APL) with PML-RARA
- AML with t(9;11)(p21.3;q23.3); MLLT3-KMT2A
- AML with t(6;9)(p23;q34.1); DEK-NUP214
- AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM
- AML (megakaryoblastic) with t(1;22)(p13.3;q13.3); RBM15-MKL1
- Provisional entity: AML with BCR-ABL1
- AML with NPM1 mutation
- AML with biallelic CEBPA mutation
- Provisional entity: AML with RUNX1 mutation
AML with myelodysplasia-associated changes
Therapy-associated myeloid neoplasia AML, NOS
- AML with minimal differentiation
- AML without maturation
- AML with maturation
- Acute myelomonocytic leukaemia
- Acute monoblastic/monocytic leukaemia
- Pure erythrocyte leukaemia
- Acute megakaryoblastic leukaemia
- Acute basophilic leukaemia
- Acute panmyelosis with myelofibrosis
Myeloid sarcoma
Myeloid Down syndrome-associated proliferation
- Transient abnormal myelopoiesis (TAM)
- Myeloid leukaemia associated with Down syndrome
Conventional cytogenetic analysis is mandatory in the evaluation of suspected AML. A bone marrow blast count of ≥20% is required for the diagnosis of AML, with the exception of AML with the recurrent genetic aberrations t(15;17), t(8;21), inv(16) or t(16;16). In addition to age, cytogenetic and molecular genetic changes, which are categorised into three groups according to the ELN classification, have a major influence on the prognosis (see table).
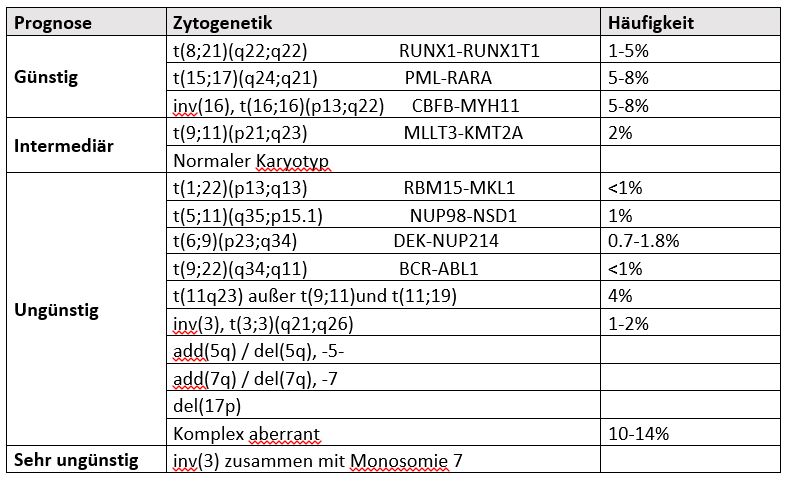
Literature:
- Onkopedia guideline „Akute myeloische Leukämie AML“ DGHO October 2019
- Döhner, Hartmut et al. “Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel.” Blood vol. 129,4 (2017): 424-447. doi:10.1182/blood-2016-08-733196
- Myeloproliferative Neoplasien (MPN)
- Myelodysplastische / myeloproliferative Erkrankungen
- Myelodysplastische Syndrome (MDS)
- Biphänotypische akute Leukämien (BAL)
Acute lymphoblastic leukaemia
- B-cell acute lymphoblastic leukaemias (B-ALL)
- T-cell acute lymphoblastic leukaemia (T-ALL)
Non-Hodgkin’s lymphoma (NHL)
Tumour cytogenetic diagnostics
-
Tumour cytogenetics deals with acquired chromosomal changes in tumours. The number and structure of chromosomes are examined using chromosome analysis methods such as banding techniques and fluorescence in situ hybridisation (FISH). Tumour cytogenetics plays a special role in haematological neoplasms, as it helps to confirm the diagnosis, has important prognostic significance, supports therapy planning and can monitor the course of the disease.
In tumour cytogenetics, acquired chromosomal abnormalities in haematological neoplasms and solid tumours are investigated. Characteristic chromosomal abnormalities that frequently occur in a certain type of tumour are associated with neoplastic transformation: they are considered primary abnormalities. Secondary abnormalities occur as the disease progresses, when the genome becomes increasingly unstable. They can contribute to the progression of the tumour.
What can tumour cytogenetics do?The cytogenetic examination of peripheral blood and bone marrow is now part of the standard diagnostics of malignant haematological diseases. It includes classic chromosome analysis together with fluorescence in situ hybridisation. Cytogenetic analysis makes an important contribution to confirming and specifying the suspected diagnosis, prognosis assessment and therapy options. It also serves to monitor the course of the disease. The identification of specific genetic markers of the malignant cell clone can also be used to monitor the course of therapy.
Pre-analytical notes and miscellaneous
-
A declaration of consent for genetic diagnostics signed by the doctor and patient must be enclosed with the request. In the case of pregnancy and prenatal diagnostics, prior agreement by telephone is required!
2 ml EDTA blood is required for analysis. These can be sent at room temperature to the following address:Labor Berlin – Charité Vivantes GmbH
Sylter Straße 2
13353 Berlin -
The GenDG stipulates that the sample must be destroyed immediately after the diagnosis has been completed. In individual cases, it may make sense to have access to a sample that is still available. For example, in the case of planned step-by-step diagnostics, possible follow-up requests, in the case of children or persons from whom a repeat blood sample could cause difficulties. If longer storage of the sample is desired, the authorisation of the patient or legal representative must be given on the declaration of consent.
The GenDG also stipulates that the findings must be deleted after 10 years. However, the patient can expressly agree to a longer storage period of up to 30 years. Knowledge of a disease-causing mutation in monogenically inherited diseases can be useful for subsequent target diagnostics in other family members.
-
The request for a genetic test is made using the referral form sample 10.
Human genetic services in accordance with Chapter 11 of the EBM (human genetics), with the exception of human genetic services in Chapter 32 of the EMB (laboratory), have no influence on the calculation of the efficiency bonus – the specification of the exception code 32010 is therefore omitted. See also: KBV
Since 1 February 2010, a declaration of consent signed by the patient and the attending physician, who must inform the patient about the nature, significance and scope of the respective examination, has been required by law for a genetic examination (part of the specific request form or separate consent form).
We cannot begin the examination without such consent. This consent can also remain with the commissioning doctor. In this case, a corresponding box must be ticked on the request form.
When the revised EBM comes into force in January 2021, the previous restriction of NGS analysis to 25kb of coding DNA will no longer apply. For patients with statutory health insurance, the use of indication-related NGS panels >25kb is now possible. Furthermore, the scope of indication-related diagnostics of monogenic diseases and their gene range has been expanded in chapter 11.4.2. Examinations in Chapter 11.4.2 are conclusive, i.e. if the suspected diagnosis could not be confirmed in a genetic examination, no further diagnostics are possible within four subsequent quarters.
The variants of unclear significance (VUS) detected in large NGS panels, which could be reassessed (reclassified) as a result of new findings, can be reassessed and diagnosed after 4 years at the earliest (a follow-up analysis is carried out exclusively using bioinformatics and does not require a new laboratory analysis).







Request forms and downloads
Services for private patients, self-paying patients or elective laboratory services shall be invoiced by LABOR BERLIN directly to the respective payer, unless otherwise agreed with the sender. For this purpose, the sender shall forward the necessary patient data to LABOR BERLIN and ensure that the patients are informed about the possible forwarding of laboratory orders to LABOR BERLIN and the associated organisational measures, including billing by a private medical clearing office, in the manner prescribed by law and consent to this. The legal requirements with regard to the free choice of doctor are taken into account. We would like to point out that, in accordance with the provisions of the Hospital Remuneration Act (KHEntgG), the ordering party must arrange for external elective laboratory services to be provided on a case-by-case basis and specifically by the elective physicians concerned.
-
Lynch syndrome request form
pdf -
Requisition slip for liver and pancreas
pdf -
Requisition slips for muscular & neuromuscular diseases
pdf -
Requisition slips for hearing impairment/deafness
pdf -
Preanalytics Molecular genetics
pdf -
Preanalytics tumor cytogenetics
pdf -
Requisition slip for prenatal cytogenetics
pdf -
Requisition slip for postnatal cytogenetics
pdf -
Requisition slip for tumor cytogenetics
pdf -
Requisition slip for cytogenetics from abortion material
pdf -
Requisition slip for connective tissue diseases
pdf -
CFTR requisition slip
pdf -
Requisition slip for endocrinological diseases
pdf -
Requisition slip for lipid metabolism disorder
pdf -
Requisition slip for cardiological diseases
pdf -
Requisition slip for bone and skeletal diseases
pdf -
Requisition slip for congenital myasthenic syndromes
pdf -
Requisition slip for lymphoedema
pdf -
Nephrogenetics requisition slip
pdf -
Noonan syndrome requisition slip
pdf -
Requisition slip for pulmonary hypertension
pdf -
Requisition slip Rett-like and Angelman syndrome
pdf -
Requisition slip for platelet disorders
pdf -
Requisition slip for primary immunodeficiencies
pdf -
Requisition slip for lung diseases
pdf