Humangenetik


Molekulargenetik und Tumorzytogenetik
Der Fachbereich Humangenetik ist aus den humangenetischen Diagnostiklaboren des Instituts für Medizinische Genetik und des Instituts für Humangenetik der Charité-Universitätsmedizin Berlin hervorgegangen. Dem Ziel von Labor Berlin, Spitzendiagnostik auf dem aktuellen Stand von Wissenschaft und Technik für unsere Patienten/innen und Einsender/innen anzubieten, sehen sich auch die Fachbereiche Molekulargenetik und Tumorzytogenetik verpflichtet. Der molekularen und tumorzytogenetischen Diagnostik kommt in der modernen Humanmedizin, die dem Anspruch einer individuellen maßgeschneiderten Behandlung gerecht werden will, eine Schlüsselrolle zu.
Wir bieten mit einem Team aus Ärzten und Naturwissenschaftlern ein umfangreiches, diagnostisches Methodenspektrum und humangenetische Expertise in den Bereichen Molekulargenetik und Tumorzytogenetik an. Ärzte und Einsender können durch benannte Ansprechpartner auf unserer Webseite eine persönliche und fachlich kompetente Beratung zu allen klinischen und diagnostischen Fragestellungen erhalten.
MOLEKULARGENETIK Leistungsspektrum / Indikationen





Bindegewebserkrankungen & Aortopathien
Zum Anforderungsschein als pdf

Knochen- & Skeletterkrankungen
Zum Anforderungsschein als pdf- Bindegewebe Hypermobilität
- Dentinogenesis Amylogenesis Imperfecta
- Distale Arthrogrypose
- Extremitätenfehlbildung
- Hochwuchs
- Idiopathischer Kleinwuchs
- Kalzium-Phosphat-Stoffwechsel-Störung
- Kraniosynostosen
- Kurzrippen-Polydaktylie-Syndrome
- Metaphysäre Dyplasie
- Multiple epiphysäre Dysplasie
- Oligodontie
- Osteopetrose/erhöhte Knochenmineraldichte-Panel
- Osteoporose/erniedrigte Knochenmineraldichte-Panel
- Spondylometaphyseale Dysplasien
- Stickler Syndrom

Endokrinologische Erkrankungen
Zum Anforderungsschein als pdf- Adipositas
- Adrenogenitales Syndrom (AGS) / kongenitale adrenale Hyperplasie (CAH)
- DSD (difference of sexual development)
- Endokrine Tumore
- Hyperinsulinismus
- Hyperkalzämie (FHH) / Hypokalzämie (ADH)
- Hyperparathyreodismus-Hypoparathyreodismus
- Intersex Panel (Differences of sexual development)
- Kallmann Syndrom (KS) / hypogonadotroper Hypogonadismus (HH)
- MODY / Neonataler Diabetes / Nicht-insulinabhängiger Diabetes mellitus (NIDDM)
- Nebenniereninsuffizienz (NNI)
- Schilddrüse / kongenitale Hypothyreose / Hyperthyreose
- Schilddrüsenkarzinom
- Wachstumshormonmangel (IGHD / CGHD / CPHD)

Entwicklungsstörungen & Epilepsie

Muskuläre & Neuromuskuläre Erkrankungen

Nierenerkrankungen
Zum Anforderungsschein als pdf- Alport Syndrom und Nephropathien vom Typ der dünnen Basalmembran
- Autosomal dominante tubuläre Nierenerkrankung
- Bartter Syndrom und Gittelman Syndrom
- CAKUT
- distale renal-tubuläre Azidose
- Hypertonie
- Joubert Syndrom
- Komplementerkrankung
- Meckel Gruber Syndrom
- Nephronophthise
- Nephrotisches Syndrom
- Nierenkarzinom
- Nierensteine
- Polycystische Nieren
- Senior Loken Syndrom, Nephronophthise mit einer Retinadegeneration

Kardiologische Erkrankungen
Zum Anforderungsschein als pdf- Arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC)
- Brugada-Syndrom (BrS)
- Catecholaminerge polymorphe ventrikuläre Tachykardie (CPVT)
- Dilatative Kardiomyopathie
- Hypertrophe Kardiomyopathie
- Long QT-Syndrom (LQTS)
- Pulmonal-arterielle Hypertonie
- Restriktive Kardiomyopathie (RCM)
- Short QT-Syndrom (SQTS)

Primäre Immundefekte
Zum Anforderungsschein als pdf- Agammaglobulinämie(PID)
- Allgemeine variable Immundefekterkrankungen (CVID)
- Autoimmun-Lymphoproliferatives Syndrom (ALPS)
- Autoinflammatorische Erkrankungen
- CD4-Lymphopenie
- Chronische mukokutane Candidiasis
- EBV assoziierte lymphoproliferative Erkrankungen
- familäres Mittelmeerfieber
- Genetische Suszeptibilität für Mykobakteriosen (MSMD)
- Hereditäres Angioödem (HAE)
- HSV1-Anfälligkeit
- Hyper-IgE-Syndrom (HIES)
- Hyper-IgM-Syndrom
- Neutropenie
- Phagozytenfunktionsdefekte (z.B. septische Granulomatose)
- schwere_COVID-19_Performanz
- Sehr früh beginnende chronisch-entzündliche Darmerkrankungen (VEO-IBD)
- T-B SCID
- T-B+ SCID

Stoffwechselerkrankungen
Zum Anforderungsschein als pdf- Aminosäuren – Transportsystem Defekte
- CDG Syndrome (basis Panel)
- CDG Syndrome (erweitertes Panel)
- Cobalamin- und Folsäure Stoffwechsel
- Eicosanoid, Glycerol, Glykophospholipid Metabolismus
- Erkrankungen des Eisenstoffwechsels
- Erkrankungen des Glutamat und Aspartat Metabolismus
- Erkrankungen des Kupferstoffwechsels und andere Metalle
- Erkrankungen des Lysin, Hydroxylysin, Tryptophan und Histidin Metabolismus
- Erkrankungen des Ornithin, Prolin, Hydroxyprolin und Oxalat Metabolismus
- Erkrankungen des Purin Stoffwechsels
- Erkrankungen des Pyrimidin Stoffwechsels
- Erkrankungen mit komplexen Moleküldegradierungen
- Fettstoffwechselstörung (HDL-Mangel)
- Fettstoffwechselstörung durch LDL-Mangel
- Fruktose und Galaktose Stoffwechsel
- Gallensäuren Metabolismus
- Glykogenspeicherkrankheiten (GSD)
- Harnstoffzyklusdefekte und angeborene Hyperammoniämie
- Hypercholesterinämie
- Hyperlipidämie (kombiniert)
- Hypertriglyceridämie
- Ketonkörper und Riboflavin Stoffwechsel
- Lipodystrophie
- mitochondriale Fettsäureoxidation und Carnitin Stoffwechsel
- Mukopolysaccharidosen und Oligosaccharidosen
- Neuronale Ceroid-Lipofuszinosen (NCL)
- Organazidurien
- Pentose und Hexose Stoffwechsel
- Peptid-, Amin- und Polyaminstoffwechse
- Peroxysmale Erkrankungen
- Phenylalanin und Tyrosin Metabolismus
- Porphyrie
- Serin und Glycin Metabolismus
- Sphingolipidosen
- Statin-assoziierte Myopathie
- Störungen der Sterolsynthese
- Störungen des Metabolismus schwefelhaltiger Aminosäuren und hydrogen Sulfide
- Störungen des Vitaminstoffwechsels – erweitertes Panel
- Synthese und Recyclingstörungen von Sphingolipiden
- verzweigtketten Krankheiten

Leber- & Pankreaserkrankungen
Zum Anforderungsschein als pdf- Cholestase – erweitertes Panel
- Cholestase – Stoffwechsel bedingt
- Cholestase – γ – GT nicht erhöht
- genetisch bedingte chronische Pankreatitis
- genetisch bedingte Hyperbilirubinämien
- genetisch bedingtes akutes Leberversagen
- hepatische Eisen- und Kupferspeicherkrankheiten
- Pankreaskarzinom
- Störungen der hepatischen Glukoneogenese
Unklare Diagnose
-
Ansprechpartner:
- Dr. rer. nat Jörg Schuldes (Joerg.Schuldes@laborberlin.com)
- Dr. rer. medic. Johannes Grünhagen (johannes.gruenhagen@laborberlin.com)
Die Untersuchung aller kodierender Abschnitte des Genoms (Whole Exome Sequencing) erlaubt eine flexible Nutzung der Daten für verschiedenste Indikationen. Von WES profitieren ganz besonders komplexe Fragestellungen, wie ungeklärte Entwicklungsstörungen, Polygene Erkrankungsbilder und besondere seltene Erkrankungen. WES bietet die Möglichkeit, auch Jahre nach erfolgter Diagnostik den Umfang der untersuchten Gene zu aktualisieren, falls auf Basis neuer wissenschaftlicher Erkenntnisse weitere Gene als ursächlich für die ursprüngliche Indikation bekannt werden oder zu erweitern, wenn sich die ursprüngliche Indikation aufgrund anderer oder weiterer klinischer Symptome ändert. Sowohl Erweiterung als auch Aktualisierung eines auf einem WES-Datensatz beruhenden virtuellen Panels kann nur nach Rücksprache erfolgen.
WES kann aufgrund dieser Tatsache flexibel bei Patienten eingesetzt werden, für deren Erkrankung keine Panel-Analyse zur Verfügung steht. Für die korrekte Auswahl von krankheitsrelevanten Genen (virtuelles Genpanel) ist eine präzise Definition klinischer Informationen über den Indexpatienten im Format von HPO-Terms (HPO: Human Phenotype Ontology; hpo.jax.org) erforderlich. Auf Basis des generierten Exom-Datensatzes erlaubt die bioinformatische Eingrenzung im Rahmen der 25kb Basisdiagnostik die Zusammenstellung spezifischer Gene, welche nach aktuellstem Stand der Wissenschaft mit dem Erkrankungsbild assoziiert sind. Dieses individuelle Panel kann nachfolgend regulär ausgewertet und befundet werden. Das Whole Exome Sequencing als Einzelanforderung stellt jedoch noch keine Regelleistung der Krankenkassen dar. Bitte kontaktieren Sie uns für weitere Informationen.
-
Patienteninformation Zusatzbefunde
pdf
Methodenspektrum Molekulardiagnostik
-
Die molekulargenetische Diagnostik ist Teil der Humangenetik, deren Ziel die Aufklärung molekularer Ursachen von genetisch bedingten Krankheiten des Menschen ist. Durch spezifische DNA-Untersuchungen bestimmter Gene können Veränderungen (Mutationen) bei Patienten identifiziert werden, die für das Auftreten einer Krankheit verantwortlich sind. Die durch die molekulargenetische Diagnostik gewonnenen Erkenntnisse sind äußerst wichtig für die weitere genetische Beratung der Familie und der darauffolgenden z.B. prädiktiven und/oder vorgeburtlichen Diagnostik. Bis heute sind ca. 14.000 Gene mit vermuteten Krankheitsassoziationen und ca. 6.000 Erkrankungen mit klar definierter molekulargenetischer Ursache bekannt (OMIM Datenbank).
Bei dem Großteil handelt es sich um sehr seltene monogene Erkrankungen, die seltener als bei 1 Fall pro 10.000 Personen in der Bevölkerung vorkommen. Von derart seltenen Krankheiten sind weltweit jedoch ungefähr 1% aller Neugeborenen betroffen.
Seltenen genetischen Erkrankungen zugrunde liegende Mutationen sind häufig noch nicht in Datenbanken beschrieben und teils schwer zu bewerten. Je länger eine Diagnostik dieser Gene erfolgt, desto mehr profitiert die Diagnostik von der Erfahrung der Auswerter und wachsenden Datenbankeinträgen. Dies erleichtert die Aufgabe, krankheitsursächliche Mutationen von Varianten ohne Krankheitswert zu unterscheiden. Die bei der Mutationssuche eingesetzten Verfahren haben sich vor allem durch den Zugang zu Populationsdaten, dem Einsatz von Bewertungsstandards (ACMG-Kriterien) und Vorhersagealgorithmen (Mutation Taster, Polyphen, DANN, FATHMM, diverse Meta-Scoring-Systeme) in den vergangenen 10 Jahren rasant weiterentwickelt.Labor Berlin bietet betroffenen Personen und Familien in Zusammenarbeit mit ärztlichen Kollegen und der genetischen Beratung eine Diagnostik auf höchstem Niveau. Um Diagnostik zum Vorteil der Betroffenen zu ermöglichen, nimmt die Humangenetik regelmäßig und erfolgreich an Ringversuchen teil, verfolgt Fortschritte und Erkenntnisse auf diesem sich schnell entwickelnden Gebiet und nutzt die neuesten Sequenziertechnologien am Markt.
Für die molekulargenetischen Analysen kommen verschiedene Methoden zum Einsatz:
- Next Generation Sequencing
- Whole Exom-Sequenzierung (WES)
- PCR (Polymerase Chain Reaction)
- Sequenzierung nach Sanger
- MLPA (Multiplex Ligation-dependent Probe Amplification)
- Quantitative Real-Time PCR
- Fragmentanalyse
-
NextGeneration Sequencing (NGS) bezeichnet eine neuartige Technologie der Nukleinsäureanalytik, mit der eine Hochdurchsatz- oder Parallelsequenzierungstechnologie gemeint ist. Im Gegensatz zur Sanger-Sequenzierung können simultan mehrere hundert Millionen Genfragmente verschiedenster Patienten in einem Prozess sequenziert werden. Das Verfahren besteht aus zwei Schritten: Im ersten Schritt wird die DNA relevanter Gene angereichert. Der zweite Schritt ist die Sequenzierung der angereicherten DNA mittels NGS.
Um die kodierenden genomischen Regionen von Interesse anzureichern, haben sich In-Solution-Capture-Methoden durchgesetzt. Eine Mischung künstlich synthetisierter lokusspezifischer Oligonukleotide (sogenannte Sonden) wird in Lösung zur fragmentierten genomischen DNA-Probe gegeben und hybridisiert dort selektiv an die genomischen Regionen von Interesse.
Die Sonden sind derart markiert, dass sie einschließlich der gebundenen DNA-Fragmente in einem nächsten Schritt physisch gebunden und gewaschen werden können, um nicht hybridisierte und überschüssige DNA-Fragmente zu entfernen. Im letzten Schritt werden die Sonden entfernt und die genomischen Fragmente können sequenziert werden.
Für die Sequenzierung nutzen wir Sequenziergeräte des Marktführers Illumina. Mithilfe einer leistungsstarken und für NGS optimierten IT-Infrastruktur werden die Rohdaten schließlich in der hauseigenen bioinformatischen GATK-basierten Pipeline aufgearbeitet und mittels spezieller Analysetools ausgewertet.Die Gendiagnostik profitiert in hohem Maße von NGS, da die Anreicherung genomischer Zielbereiche eine höhere diagnostische Sensitivität, die parallele Untersuchung von Genen in so gennannten „Genpanels“ zeitsparend ist und eine kostengünstigere Bearbeitung vieler Proben erlaubt. NGS hat sich daher als Standardsequenziertechnologie etabliert, die wir in der Humangenetik für ein breites Spektrum an Fragestellungen im diagnostischen Bereich nutzen (siehe Diagnostikpanels).
-
-
Die Exomsequenzierung, auch bekannt als Whole Exom-Sequenzierung (WES), ist eine genomische Technik zur Sequenzierung aller kodierenden Regionen („Exons“) in einem Genom (“Exom“). Das Verfahren besteht aus zwei Schritten: Im ersten Schritt wird die exonische DNA angereichert. Die exonische DNA des Menschen besteht aus ~180.000 Exons, die etwa 1-2% des menschlichen Genoms ausmachen (50 Millionen Basenpaare). Der zweite Schritt ist die Sequenzierung der exonischen DNA mittels NGS (Beschreibung im Abschnitt Next Generation Sequencing (NGS))
Um die kodierenden genomischen Regionen von Interesse anzureichern, haben sich In-Solution-Capture-Methoden durchgesetzt. Ein Pool künstlich synthetisierter lokusspezifischer Oligonukleotide (sogenannte Sonden) wird in Lösung zur fragmentierten genomischen DNA-Probe gegeben und hybridisiert dort selektiv an die genomischen Regionen von Interesse.
Die Sonden sind derart markiert, dass sie einschließlich der gebundenen DNA-Fragmente in einem nächsten Schritt physisch gebunden und gewaschen werden können, um nicht hybridisierte und überschüssige DNA-Fragmente zu entfernen. Im letzten Schritt werden die Sonden entfernt und die genomischen Fragmente können sequenziert werden.
Die Whole Exom-Sequenzierung-Diagnostik stellt insbesondere bei Patienten mit komplexen Fragestellungen oder unspezifischer Symptomatik eine sinnvolle Methodik dar, da hier oft kein geeignetes Gen-Panel zur Aufklärung der genetische Ursache zur Verfügung steht. Nach oft jahrelanger erfolgloser Diagnostik kann die Whole Exom-Sequenzierung das Mittel der Wahl sein, um eine genetische Ursache für die Erkrankung zu identifizieren.
-
PCR (Polymerase Chain Reaction; deutsch Polymerase Kettenreaktion)
Anwendung in der Diagnostik: Teil der NGS-Methodik, Teil der Sanger-Sequenzierung, Teil der MLPA
Die PCR (engl. Abkürzung für polymerase chain reaction) bezeichnet eine Methode, um Teile der Erbsubstanz DNA in vitro, das bedeutet außerhalb eines biologischen Systems, zu vervielfältigen. PCR wird eingesetzt, um einen oder mehrere kurze, genau definierte Teile einer Ausgangs-DNA zu vervielfältigen. Dabei kann es sich bei dem zu vervielfältigenden Abschnitt um ein Gen oder den Teilabschnitt eines Gens handeln oder auch um nichtcodierende DNA-Sequenzen. Für die PCR bedient man sich des Enzyms DNA-Polymerase, welches in lebenden Zellen ebenfalls für die DNA-Vervielfältigung, also das Kopieren der Erbinformation essentiell ist.
Die PCR benötigt mehrere Komponenten.
- Die Original-DNA als Template
- Zwei oder mehr Primer
- Eine hitzebeständige DNA-Polymerase
- Desoxyribonucleosidtriphosphate, die Grundbausteine der DNA
- MG2+ Ionen, ohne die eine Funktion der Polymerase nicht gewährleistet ist
- Eine Pufferlösung, die eine möglichst physiologische Reaktionsumgebung für die Polymerase sicherstellt.
Der Begriff Polymerase-Kettenreaktion beschreibt nun den Vorgang eines sich wiederholenden Programms zur Vervielfältigung der DNA. Dieses zyklische Programm setzt auf eine thermische Destabilisierung des DNA-Doppelstranges bei >95°C und eine folgend stattfindende Verdopplung des DNA-Stranges bei ca. 60°C. Eine Kettenreaktion findet in diesem Zusammenhang aufgrund der Tatsache statt, dass die Produkte eines Zyklus die Ausgangsstoffe (Templates) für den nächsten Zyklus darstellen und somit eine exponentielle Vervielfältigung stattfinden kann.
Die PCR findet als Basismethodik bei der Sanger-Sequenzierung, der MLPA, NGS, der Quantitativen PCR, der Fragmentanalyse und dem Methylierungstest Anwendung.
-
Die Sanger-Sequenzierung bzw. Didesoxymethode nach Sanger ist eine nach Frederick Sanger, einem englischen Biochemiker benannte Methode der DNA-Sequenzbestimmung. Sie beruht auf dem Prinzip der Kettenabbruchmethode und erlaubt die Bestimmung der Basenabfolge in einem spezifischen DNA-Molekül. Es können nicht zwei verschiedene DNA-Abschnitte in einem Prozess analysiert werden.
Das Prinzip der DNA-Sequenzierung nach Sanger besteht in der Synthese neuer DNA-Stränge unterschiedlicher Länge, die anschließend in einem Kapillarsequenzierer nach Größe getrennt die Sequenzinformation bestimmbar macht. Um DNA-Stränge unterschiedlicher Länge zu erzeugen, werden neben dem Nukleotid-Mix zusätzlich fluoreszenzmarkierte Stoppnukleotide (Dideoxy-Nukleotide) eingesetzt. Für jedes der vier möglichen Nukleotide Adenin, Guanin, Cytosin und Thymin existiert ein äquivalentes, spezifisch fluoreszenzfarbstoffmarkiertes Stoppnukleotid. Durch die richtige Mischung von Nukleotid-Mix und Stoppnukleotiden entsteht in einer PCR theoretisch jedes denkbar lange Sequenzfragment.
Durch die je nach Nukleotid verschieden fluoreszenzmarkierten Fragmente kann nach Größenauftrennung eine Auswertung der Basenabfolge mit Hilfe einer speziellen Software vorgenommen werden.
Wir setzen die Sanger-Sequenzierung in der Routine zum Nachweis bekannter familiärer Mutationen und bei der Bestätigung fraglicher NGS-Varianten ein.
-
Das menschliche diploide Genom weist, bis auf die Geschlechtschromosomen, ein paarweises Vorkommen der Chromosomen auf. Auf jedem dieser Chromosomenpaare liegen im Normalfall jeweils die gleichen Gene einmal vor, man spricht auch von zwei Allelen (oder bi-allelisch). Nun kann es durch Vorgänge während der Meiose oder Mitose zu ungleichen Verteilungen der DNA-Abschnitte und damit zu einem Verlust oder Zugewinn von DNA-Material auf den Chromosomen kommen. Man spricht hier auch von Gendosisunterschieden im Vergleich zum Normalzustand, dem Wildtyp. Diese Gendosisunterschiede lassen sich mit der herkömmlichen PCR-Methode normalerweise nicht nachweisen, da die Kettenreaktion exponentiell vervielfältigt und anfängliche Dosisunterschiede zum Ende der Vervielfältigungsphase verwischen.
Mit der MLPA-Methode lassen sich Dosisunterschiede von bis zu 50 verschiedenen Nukleinsäurefragmenten in einem Ansatz detektieren. Sie wurde erstmals 2002 beschrieben (Schouten et al., Nucl Acids Res 30:12e57) und basiert auf der Ligation von zwei Lokus-spezifischen Sonden auf der DNA, die dosisabhängig zu einem Fragment verbunden werden.
Die Menge der sequenzspezifisch hybridisierten und ligierten Sonden ist somit proportional zur Kopienzahl der Ziel-Sequenz.
Es schließt sich eine PCR und eine kapillarelektrophoretische Trennung der Produkte nach Größe an. Die Fragmente können durch ihre definierte unterschiedliche Größe einem spezifischen Lokus zugeordnet werden.
Dosisunterschiede sind durch Reduktion oder Vergrößerung der Peakhöhen und -flächen im Vergleich zu den Referenzproben berechenbar.
Das Angebot an MLPA-Kits durch den Hersteller MRC-Holland ist mittlerweile sehr umfangreich. So sind MLPA-Kits zur Detektion von Deletionen und Duplikationen vielfältiger genomischer Bereiche im Rahmen verschiedenster klinischer Fragestellungen kommerziell verfügbar. Eine Erweiterung der klassischen MLPA stellen z.B. methylierungsspezifische MLPAs (MS-MLPA) dar, die sowohl zur Bestimmung der Kopienzahl als auch zur Analyse des Methylierungsmusters (Detektion von Imprinting-Defekten) und zur Analyse von Tumoren eingesetzt werden können.
Eine Alternative zur Bestimmung der Gendosis stellt die Quantitative Real-Time PCR dar (siehe dort).
-
-
Die quantitative Real-Time-PCR ist eine Vervielfältigungsmethode für Nukleinsäuren, die auf dem Prinzip der herkömmlichen Polymerase-Kettenreaktion (PCR) beruht. Sie ist jedoch zusätzlich mit einer Fluoreszenz-Messung in der exponentiellen Phase der PCR gekoppelt und ermöglicht so die Quantifizierung von Genabschnitten oder Transkripten. Wir setzen bei unserer Diagnostik auf ein Gerät von Life Technologies. In diesem Gerät findet die Detektion von PCR-Produkten entweder sequenzunspezifisch statt (z.B. über SYBR Green) oder sequenzspezifisch mittels fluoreszenzmarkierter Sonden.
Die Zunahme des Fluoreszenzsignals im Verlauf der Reaktion ist proportional zur Menge des entstehenden PCR-Produkts und kann im Verlauf der Reaktion in Echtzeit verfolgt werden.
Die Dosis des Ausgangmaterials kann somit absolut oder relativ zu einer internen Kontrollsequenz und externen Kontrollprobe gemessen werden. Der Vorteil gegenüber käuflichen Kits zur Dosisbestimmung liegt in der Flexibilität. Sequenzspezifische Primer lassen sich für beliebige Loci im Genom herstellen.
Ein Nachteil dieses Verfahrens ist die geringe Spezifität, da zwischen gewünschten und nicht erwünschten PCR-Produkten nicht unterschieden werden kann. Parallele Messungen mehrerer Loci (Multiplex-Messungen) können ebenfalls nicht durchgeführt werden. Den ersten Nachteil gleichen wir durch die Durchführung einer Schmelzkurvenanalyse aus, anhand derer die Fragmentlänge(n) und dadurch die Spezifität bestimmt werden kann.
Eine Alternative zur Bestimmung der Gendosis stellt die MLPA dar (siehe dort).
-
Die Fragmentanalyse beruht auf dem Funktionsprinzip der Polymerase Kettenreaktion (PCR). Im Gegensatz zur klassischen PCR werden für die Fragmentanalyse sequenzspezifische fluoreszensmarkierte Primerpaare eingesetzt. Durch Hinzufügen eines fluoreszenzmarkierten Längenstandards und Auftrennung dieser markierten PCR-Produkte nach Größe in einem Kapillarsequenzierer ist im Gegensatz zur Gelelektrophorese eine Größenunterscheidung von nur einer Base möglich. Durch die Verwendung mehrerer unterschiedlicher Fluoreszenzfarbstoffe können in einem PCR-Ansatz parallel dreistellige Zielsequenzen analysiert werden.
Zum Einsatz kommt die Fragmentanalyse bei der Chimärismusanalytik, Monosomiediagnostik, Untersuchungen auf Chromosomenaberrationen, Vaterschaftstests und Abstammungsuntersuchungen.



TUMORZYTOGENETIK
Leistungsspektrum / Indikation
-
Akute myeloische Leukämie (AML)
Die Akute myeloische Leukämie ist eine heterogene Erkrankung, die de novo entstehen, therapieassoziiert (t-AML) sein oder sekundär aus einer vorbestehenden myeloproliferativen Erkrankung bzw. einem MDS (s-AML) hervorgehen kann. Die Inzidenz steigt mit dem Alter an und beträgt ca. 3,7 Erkrankungen/100.000 Einwohner pro Jahr, das mediane Alter liegt bei 72 Jahren.
Gemäß WHO Klassifikation 2017 wir die AML in folgende Subgruppen unterteilt:
Akute myeloische Leukämien (AML) und verwandte Neoplasien
AML mit rekurrenten genetischen Aberrationen
- AML mit t(8;21)(q22;q22.1); RUNX1-RUNX1T1
- AML mit inv(16)(p13.1q22) oder t(16;16)(p13.1;q22); CBFB-MYH11
- Akute Promyelozytenleukämie (APL) mit PML-RARA
- AML mit t(9;11)(p21.3;q23.3); MLLT3-KMT2A
- AML mit t(6;9)(p23;q34.1); DEK-NUP214
- AML mit inv(3)(q21.3q26.2) oder t(3;3)(q21.3;q26.2); GATA2, MECOM
- AML (megakaryoblastisch) mit t(1;22)(p13.3;q13.3); RBM15-MKL1
- Provisorische Entität: AML mit BCR-ABL1
- AML mit NPM1-Mutation
- AML mit biallelischer CEBPA-Mutation
- Provisorische Entität: AML mit RUNX1-Mutation
AML mit Myelodysplasie-assoziierten Veränderungen
Therapie-assoziierte myeloische Neoplasie AML, NOS
- AML mit minimaler Differenzierung
- AML ohne Ausreifung
- AML mit Ausreifung
- Akute myelomonozytäre Leukämie
- Akute Monoblasten-/Monozyten-Leukämie
- Reine Erythozytenleukämie
- Akute Megakaryoblastenleukämie
- Akute Basophilenleukämie
- Akute Panmyelose mit Myelofibrose
Myeloisches Sarkom
Myeloische Down-Syndrom-assoziierte Profliferation
- Transiente abnormale Myelopoese (TAM)
- Myeloische Leukämie assoziiert mit Down-Syndrom
Die konventionelle zytogenetische Analyse ist obligatorisch bei der Beurteilung eines Verdachts auf AML. Für die Diagnose einer AML ist eine Knochenmarkblastenzahl von ≥20% erforderlich, mit Ausnahme der AML mit den rekurrenten genetischen Aberrationen t(15;17), t(8;21), inv(16) oder t(16;16). Einen großen Einfluss auf die Prognose haben neben dem Alter die zytogenetischen und molekulargenetischen Veränderungen, die nach ELN-Klassifikation in drei Gruppen eingeteilt werden (siehe Tabelle).
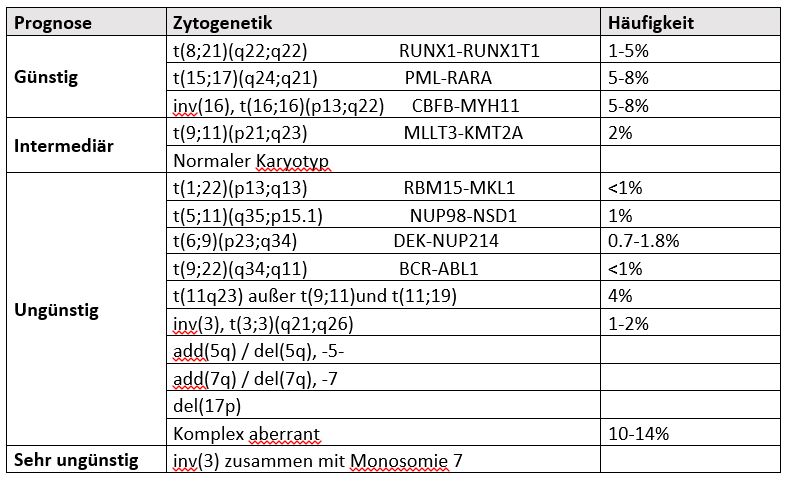
Literatur:
- Onkopedia Leitlinie „Akute myeloische Leukämie AML“ DGHO Oktober 2019
- Döhner, Hartmut et al. “Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel.” Blood vol. 129,4 (2017): 424-447. doi:10.1182/blood-2016-08-733196
- Myeloproliferative Neoplasien (MPN)
- Myelodysplastische / myeloproliferative Erkrankungen
- Myelodysplastische Syndrome (MDS)
- Biphänotypische akute Leukämien (BAL)
Akute lymphatische Leukämien
- B-Zell akute lymphatische Leukämien (B-ALL)
- T-Zell akute lymphatische Leukämien (T-ALL)
Non-Hodgkin-Lymphome (NHL)
Tumorzytogenetische Diagnostik
-
Die Tumorzytogenetik befasst sich mit erworbenen Chromosomenveränderungen in Tumoren. Dabei werden die Anzahl und die Struktur der Chromosomen durch die Methoden der Chromosomenanalyse mittels Bänderungstechniken und Fluoreszenz-in-situ-Hybridisierung (FISH) untersucht. Eine besondere Rolle spielt die Tumorzytogenetik bei hämatologischen Neoplasien, da sie zur Sicherung der Diagnose beiträgt, eine wichtige prognostische Bedeutung hat, bei der Therapieplanung unterstützt sowie Krankheitsverläufe kontrollieren kann.
In der Tumorzytogenetik werden erworbene Chromosomenanomalien bei hämatologischen Neoplasien und soliden Tumoren untersucht. Charakteristische Chromosomenanomalien, die häufig bei einem bestimmten Tumortyp auftreten, werden mit der neoplastischen Transformation assoziiert: Sie gelten als primäre Anomalien. Sekundäre Anomalien treten beim Fortschreiten der Erkrankung auf, wenn das Genom zunehmend instabil wird. Sie können zur Progression des Tumors beitragen.
Was kann die Tumorzytogenetik leisten?Die zytogenetische Untersuchung von peripherem Blut und Knochenmark gehört heute zur Standarddiagnostik maligner hämatologischer Erkrankungen. Sie umfasst die klassische Chromosomenanalyse zusammen mit der Fluoreszenz-in-situ-Hybridisierung. Die zytogenetische Analyse leistet einen wichtigen Beitrag, die Verdachtsdiagnose, die Prognoseabschätzung, die Therapie-Optionen zu sichern und zu spezifizieren. Außerdem dient sie zur Kontrolle des Krankheitsverlaufs. Die Identifizierung spezifischer, genetischer Marker des malignen Zellklons kann außerdem dazu herangezogen werden, den Therapie-Verlauf zu überwachen.
Präanalytische Hinweise und Sonstiges
-
Eine von Arzt und Patient unterschriebene Einverständniserklärung zur genetischen Diagnostik ist der Anforderung beizulegen. Bei Schwangerschaft und pränataler Diagnostik ist eine vorherige telefonische Vereinbarung erforderlich!
Zur Analyse werden 2 ml EDTA Blut benötigt. Diese können bei Raumtemperatur an die folgende Adresse gesendet werden:Labor Berlin – Charité Vivantes GmbH
Sylter Straße 2
13353 Berlin -
Das GenDG schreibt nach Abschluss der Diagnostik eine sofortige Vernichtung der Probe vor. Es kann in Einzelfällen sinnvoll sein, auf eine noch vorhandene Probe Zugriff zu haben. So z.B. bei geplanter Stufendiagnostik, eventuellen Nachforderungen, bei Kindern oder Personen, bei denen eine erneute Blutentnahme Schwierigkeiten bereiten könnte. Ist eine längere Aufbewahrung der Probe gewünscht, muss die Erlaubnis des Patienten bzw. des gesetzlichen Vertreters auf der Einwilligungserklärung erteilt werden.
Das GenDG schreibt ferner die Löschung des Befunds nach 10 Jahren vor. Der Patient kann aber ausdrücklich einer längeren Aufbewahrung von bis zu 30 Jahren zustimmen. Die Kenntnis einer krankheitsverursachenden Mutation bei monogen vererbten Erkrankungen kann für die spätere Zieldiagnostik bei weiteren Familienmitgliedern sinnvoll sein.
-
Die Anforderung einer genetischen Untersuchung erfolgt über den Überweisungsschein Muster 10.
Humangenetische Leistungen nach Kapitel 11 des EBM (Humangenetik), mit Ausnahme der humangenetischen Leistungen des Kapitel 32 des EMB (Labor), haben auf die Ermittlung des Wirtschaftlichkeitsbonus keinen Einfluss – die Angabe der Ausnahmekennziffer 32010 entfällt daher. Siehe hierzu auch: KBV
Seit dem 01.02.2010 ist für eine genetische Untersuchung eine vom Patienten und behandelndem Arzt, der den Patienten über Wesen, Bedeutung und Tragweite der jeweiligen Untersuchung aufzuklären hat, unterschriebene Einverständniserklärung gesetzlich vorgeschrieben (Teil der spezifischen Anforderungsscheine bzw. separate Einwilligungsformular).
Ohne eine solche Einwilligung können wir nicht mit der Untersuchung beginnen. Diese Einwilligung kann auch beim beauftragenden Arzt verbleiben. In diesem Fall ist auf dem Anforderungsschein ein entsprechendes Feld zu markieren.
Mit Inkrafttreten des überarbeiteten EBM im Januar 2021 entfällt die vormalige Beschränkung der NGS-Analytik auf 25kb kodierender DNA. Für gesetzlich krankenversicherte Patienten ist ab sofort der Einsatz von indikationsbezogenen NGS-Panels >25kb möglich. Weiterhin wurde der Umfang indikationsbezogener Diagnostik monogener Erkrankungen und deren Genumfang in Kapitel 11.4.2 erweitert. Untersuchungen in Kapitel 11.4.2 sind abschließend, d.h. konnte bei einer genetischen Untersuchung die Verdachtsdiagnose nicht bestätigt werden, ist innerhalb von vier Folgequartalen keine weitere Diagnostik möglich.
Die bei großen NGS-Panels nachgewiesenen Varianten unklarer Signifikanz (VUS), die durch neue Erkenntnisse eine Neubewertung (Reklassifikation) erhalten könnten, sind frühestens nach 4 Jahren erneut beurteil- und befundbar (Eine Nachanalyse erfolgt ausschließlich bioinformatisch und erfordert keine erneute Laboranalytik).







Anforderungsscheine und Downloads
Leistungen für Privatpatienten, selbstzahlende Patienten oder laborärztliche Wahlleistungen werden durch LABOR BERLIN direkt mit dem betreffenden Kostenträger abgerechnet, sofern mit dem Einsender nicht etwas anderes vereinbart ist. Hierzu wird der Einsender die notwendigen Daten der Patienten an LABOR BERLIN weiterleiten und sicherstellen, dass die Patienten über die mögliche Weiterleitung von Laboraufträgen an LABOR BERLIN und die damit verbundenen organisatorischen Maßnahmen, einschließlich der Abrechnung durch eine privatärztliche Verrechnungsstelle, in der gesetzlich vorgeschriebenen Weise informiert werden und hierin einwilligen. Die rechtlichen Vorgaben in Bezug auf die freie Arztwahl werden dabei berücksichtigt. Wir weisen darauf hin, dass die Veranlassung externer laborärztlicher Wahlleistungen nach den Regelungen des Krankenhausentgeltgesetzes (KHEntgG) durch den Einsender einzelfallbezogen und konkret durch die betreffenden Wahlärzte zu erfolgen hat.
-
Anforderungsschein Lynch-Syndrom
pdf -
Anforderungsschein Leber und Pankreas
pdf -
Anforderungsschein Muskuläre & Neuromuskuläre Erkrankungen
pdf -
Anforderungsschein Hörstörung/Taubheit
pdf -
Präanalytik Molekulargenetik
pdf -
Patienteninformation Zusatzbefunde
pdf -
Präanalytik Tumorzytogenetik
pdf -
Anforderungsschein pränatale Zytogenetik
pdf -
Anforderungsschein postnatale Zytogenetik
pdf -
Anforderungsschein Tumorzytogenetik
pdf -
Anforderungsschein Zytogenetik aus Abortmaterial
pdf -
Anforderungsschein Bindegewebserkrankungen
pdf -
Anforderungsschein CFTR
pdf -
Anforderungsschein endokrinologische Erkankungen
pdf -
Anforderungsschein Fettstoffwechselstörung
pdf -
Anforderungsschein kardiologische Erkankungen
pdf -
Anforderungsschein Knochen- und Skeletterkrankungen
pdf -
Anforderungsschein Kongenitale Myasthene Syndrome
pdf -
Anforderungsschein Lymphödem
pdf -
Anforderungsschein Nephrogenetik
pdf -
Anforderungsschein Noonan Syndrom
pdf -
Anforderungsschein Pulmonale Hypertonie
pdf -
Anforderungsschein Rett Rett-like und Angelman Syndrom
pdf -
Anforderungsschein Thrombozytenstörungen
pdf -
Anforderungsschein primäre Immundefekte
pdf -
Anforderungsschein Lungenerkrankungen
pdf -
Patienteninformation Beispiel Zusatzbefund bei WES
pdf