Hämatologie & Onkologie


Hämatologie & Onkologie
Der Fachbereich Hämatologie & Onkologie bei Labor Berlin entstand in den Jahren 2010/2011 durch Fusion der hämatologisch-onkologischen Diagnostiklabore der beiden Charité-Standorte Campus Benjamin Franklin und Campus Virchow-Klinikum mit der hämatologischen Spezialdiagnostik der Vivantes Kliniken.
Diagnostik in der Hämato-Onkologie
Krebserkrankungen zählen neben den kardiovaskulären Erkrankungen zu den führenden Todesursachen und stellen auch eine große gesundheitspolitische Herausforderung dar. In kaum einer anderen Teildisziplin der Medizin haben die Erkenntnisse der Grundlagenforschung aus den Bereichen Immunologie und Genetik so unmittelbare Anwendung gefunden wie in der Hämato-Onkologie. In diesem Gebiet wurden beispielsweise die ersten therapeutischen monoklonalen Antikörper eingesetzt und die ersten molekular-zielgerichteten Therapien (targeted therapies) entwickelt. In keinem anderen Gebiet der modernen Medizin ist die genetisch-immunologische Diagnostik so verfeinert wie beispielsweise bei den akuten Leukämien.
Tumorerkrankungen sind meist außerordentlich heterogen. Zur genauen Diagnosestellung ist daher häufig ein umfassendes diagnostisches Instrumentarium notwendig. Auch heute noch – obwohl in ihren Grundzügen zum Teil schon mehr als 100 Jahre alt – bildet die Zytomorphologie einen Grundpfeiler der hämatologischen Diagnostik. Hinzu kommen immunologische Methoden wie die Durchflusszytometrie (Immunphänotypisierung) und genetische Methoden wie die Zytogenetik und Molekulargenetik. Bei vielen hämatologischen Erkrankungen/Neoplasien kann eine sichere Diagnose nur in Zusammenschau aller dieser diagnostischen Befunde gestellt werden.
Zudem haben einige diagnostische Merkmale (immunologisch oder genetisch) eine prognostische Implikation. Im Rahmen großer Therapiestudien konnten beispielsweise bei vielen hämatologischen Erkrankungen Hoch- und Niedrigrisiko-Merkmale charakterisiert werden. Die Identifikation dieser Merkmale ist oft von Bedeutung, da die Therapie danach gesteuert werden kann.
Diagnostik
-
Auch in der modernen Hämatologie ist die Zytomorphologie ein Grundpfeiler der Diagnostik. Je nach Fragestellung wird der morphologische Befund ergänzt durch Immunzytologie (Durchflusszytometrie), Zytogenetik und Molekularbiologie.
Im Labor für Zytologie werden neben manuellen Differentialblutbildern vorwiegend Knochenmarksausstriche sowie zytologische Zentrifugenpräparate aus Liquor und aus Ergussflüssigkeiten (Pleuraerguss, Aszites, u. a.) angefertigt. Dabei kommen neben der konventionellen zytologischen Beurteilung auch zytochemische und immunzytologische Verfahren zum Einsatz. Ziel bei allen Einsendungen ist es, den Befund zügig zu erstellen: bei rechtzeitigem Probeneingang und Mitteilung einer besonderen Dringlichkeit auch noch am gleichen Tag!
Diese Untersuchungen werden angeboten:
- Manuelles Differentialblutbild, ggf. nach Leukozytenanreicherung
- Knochenmarkzytologie, ggf. inklusive Zytochemie, Eisenfärbung und Immunzytologie
- Zytologie aus Ergüssen (i. b. Pleuraerguss, Aszites), ggf. inklusive Immunzytologie
- Liquorzytologie, ggf. inklusive Immunzytologie
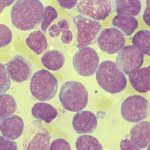
Knochenmarkaustausch einer Akuten myeloischen Leukämie (AML)
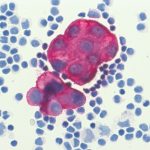
Immunzytologische Färbung: Karzinomzellen im Pleuraerguss
Die Verarbeitung der Proben sowie die Befunderstellung erfolgen werktags von Montag bis Freitag. Als Antikoagulanz muss EDTA (oder Zitrat) verwendet werden. Die Qualität der Befunde und deren Interpretation hängen wesentlich von Alter und Qualität des Probenmaterials sowie von begleitenden klinischen Daten ab. Deshalb sind schnelle Transportwege zu empfehlen. Die wichtigen klinischen Befunde sollen komplett übermittelt werden. Für die Knochenmarkzytologie ist insbesondere ein aktuelles Blutbild mit Differentialblutbild wichtig. Für eine telefonische Diskussion der Befunde steht das Laborteam gerne zur Verfügung.
Präanalytische Hinweise – Zytologie
Antikoagulanz für alle zytologischen Untersuchungen ist grundsätzlich EDTA. In Ausnahmefällen geht auch Citrat. Heparin ist dagegen ungünstig und sollte nicht verwendet werden.
Als Untersuchungsmaterial sind geeignet:
- Knochenmarkaspirat (5 ml)
- peripheres Blut (Routine-EDTA-Blutröhrchen)
- Ergussmaterial (Aszites, Pleuraerguss, Perikarderguss u.a.)
- Liquor
Es sollte darauf geachtet werden, dass die Transportzeit nicht mehr als 24 Stunden beträgt und dass das Material vor Wochenenden spätestens bis Freitag, 14.00 Uhr im Labor eintrifft. Grundsätzlich sollte Material mit einem Express-Service (Übernacht-Service) verschickt werden, um zu lange Transportzeiten zu vermeiden.
-
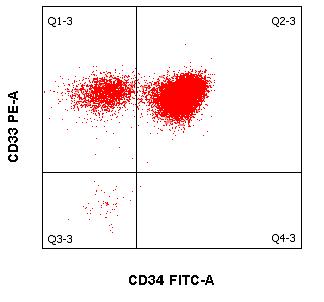
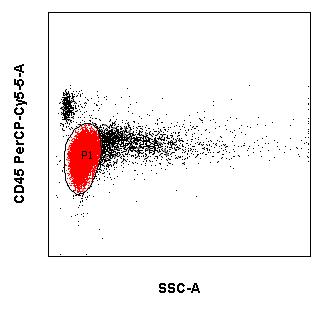
Die immunzytologische Diagnostik (mittels Durchflusszytometrie) ist ein wichtiger Eckpfeiler für Diagnostik und Verlaufsbeurteilung von Leukämien und Lymphomen neben der Morphologie, Zytogenetik und Molekularbiologie. Die Immunphänotypisierung maligner Zellen erfolgt meistens in peripherem Blut und Knochenmark. Es können jedoch auch Körperflüssigkeiten wie Liquor und Ergüsse als Untersuchungsmaterial versandt werden. Die moderne multiparametrische Durchflusszytometrie beruht wesentlich auf Fortschritten in drei Bereichen:
- Laseroptik
- Computergestützte Datenverarbeitung
- Entwicklung neuer Fluoreszenzfarbstoffe für die Kopplung an die entsprechenden monoklonalen Antikörper
Wenn mehrere Fluoreszenzfarbstoff-gekoppelte Antikörper kombiniert und die unterschiedlichen Streulichteigenschaften von Zellen ausgenutzt werden, gelingt es:
- maligne hämatologische Neoplasien zu klassifizieren und
- ggf. den Therapieerfolg im Rahmen einer Remissionsbeurteilung und der Kontrolle von minimaler Resterkrankung (MRD) zu beurteilen.
Ziel bei allen Einsendungen ist eine rasche Befunderstellung: bei rechtzeitigem Probeneingang am gleichen Tag, als Fax verschickt.
Im Einzelnen werden im Labor folgende Untersuchungen durchgeführt:
- Immunphänotypisierung von Leukämien und Lymphomen (5-10ml EDTA-Blut oder –KM)
- Remissionsbeurteilung inkl. Detektion von MRD(minimal residual disease) bei Leukämien und Lymphomen (5-10ml EDTA-Blut oder –KM)
- Detektion von Leukämie-/Lymphomzellen in Liquor und Ergussflüssigkeiten (5ml mit EDTA antikoaguliertes Probenmaterial)
- Detektion von Myelomzellen (5ml EDTA Blut oder KM)
- PNH-Diagnostik (10ml EDTA Blut)
- Quantifizierung von CD34+ hämatopoetischen Stammzellen (2ml EDTA Blut oder Stammzellasservat)
-
EMA-Test bei V.a.Sphärozytose (2ml EDTA Blut)
Haarzell-Leukämie (peripheres Blut)
Akute myeloische Leukämie (Knochenmark)
Die Verarbeitung der Proben sowie die Befunderstellung erfolgen werktags von Montag bis Freitag. Die Qualität der Befunde und deren Interpretation hängen wesentlich von Alter und Qualität des Probenmaterials sowie von begleitenden klinischen Angaben ab. Deshalb sind schnelle Transportwege zu empfehlen. Die wichtigen klinischen Befunde inkl. Blutbild und Differentialblutbild sollen komplett übermittelt werden. Das Laborteam steht gerne für eine telefonische Diskussion der Befunde zur Verfügung.
Präanalytische Hinweise – Durchflusszytometrie
Antikoagulans für alle durchflusszytometrischen Untersuchungen ist grundsätzlich EDTA. Heparin ist weniger gut geeignet und sollte nur dann verwendet werden, wenn nichts anderes verfügbar ist (z.B. versehentliche Abnahme in einer falschen Spritze bei einer Knochenmarkpunktion).
Als Untersuchungsmaterial sind geeignet:
- Knochenmarkaspirat (5 – 10 ml)
- peripheres Blut ( 5 – 10 ml)
- Ergussmaterial (Aszites, Pleuraerguss, Perikarderguss u.a.), wenn möglich 10 ml
- Liquor
- PNH-Diagnostik erfolgt aus peripherem Blut !
Der Probenversand sollte unbedingt mit einem Express-Service (Übernacht-Service) erfolgen, um die Transportzeiten so gering wie möglich zu halten. Bei Eingang im Labor sollten die Proben nicht älter als 24 Stunden sein. Vor Wochenenden ist darauf zu achten, dass die Probe spätestens Freitagmittag, um 12.00 Uhr im Labor eintrifft.
-
Die molekulare Tumorgenetik beschäftigt sich mit der Analyse molekularer Veränderungen bei malignen Erkrankungen. Das Wissen um die molekularen Veränderungen bei bösartigen Erkrankungen ist in den letzten Jahrzehnten exponentiell angewachsen, insbesondere seit der ersten Komplettsequenzierung des menschlichen Genoms 2001 im Rahmen des Humangenomprojekts. Dementsprechend haben sich auch die diagnostischen Möglichkeiten stark erweitert.
Die wichtigsten diagnostischen Methoden im Bereich der molekularen Tumorgenetik sind die verschiedenen Techniken der Polymerase-Kettenreaktion (PCR = polymerase chain reaction) und die DNA-Sequenzierung. Die molekulardiagnostische Tumorgenetik hat zwei hauptsächliche Anwendungsfelder:
- Die Erfassung molekularer Veränderungen in Tumoren. Dazu gehören Chromosomentranslokationen oder Mutationen im weitesten Sinne. Bei einigen Chromosomentranslokationen werden chimäre Fusionsgene gebildet, die als mRNA transkribiert werden. Diese chimäre mRNA kann mittels RT-PCR nachgewiesen werden. Punktmutationen können zu Aminosäure-Austauschen und damit zur Funktionsveränderung von Genen führen. Andere Mutationen führen zu Leserahmenverschiebungen, zum Verlust von Spleißstellen, etc. und damit zum Funktionsverlust des Gens.Viele dieser molekularen Veränderungen in Tumoren sind von prognostischer oder therapeutischer Bedeutung.
- Verlaufsuntersuchungen anhand molekularer Marker. Bei vielen hämatologischen Erkrankungen liegen molekulare Veränderungen vor, die sich mit einer quantitativen PCR quantifizieren lassen. Durch regelmäßige Quantifizierung dieser Veränderungen im Krankheitsverlauf lassen sich wertvolle Informationen über das Ausmaß der Erkrankung gewinnen, z. B. lässt sich die Wirksamkeit einer Therapie beurteilen. Besondere Bedeutung haben diese Messungen bei den akuten und chronischen Leukämien (MRD = minimal residual disease).
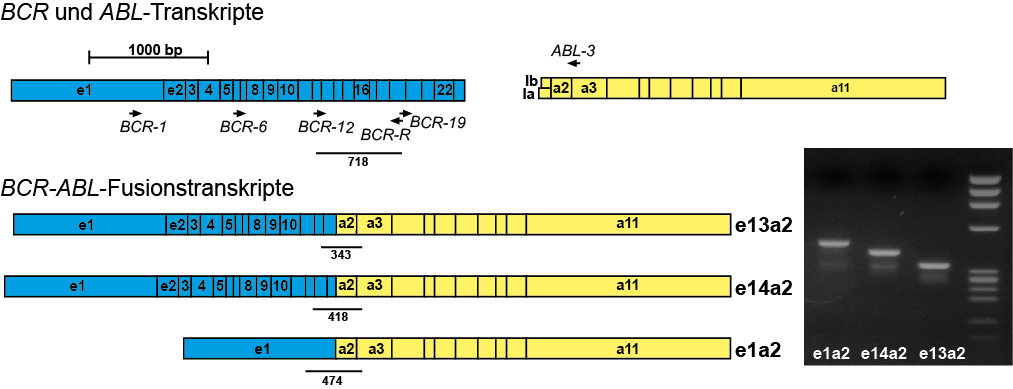
Beispiel: Nachweis der BCR-ABL-Fusion mittels RT-PCR
Methodenspektrum
Die wichtigsten molekulargenetischen Methoden, die im Labor Berlin angewendet werden, sind:
- Konventionelle PCR und RT-PCR (Detektion im Agarose-Gel)
- Langstrecken-PCR
- real-time quantitative PCR (genomische DNA oder cDNA)
- DNA-Sequenzierung (Sanger und Next generation sequencing NGS
- Chimärismusanalysen nach allogener Transplantation
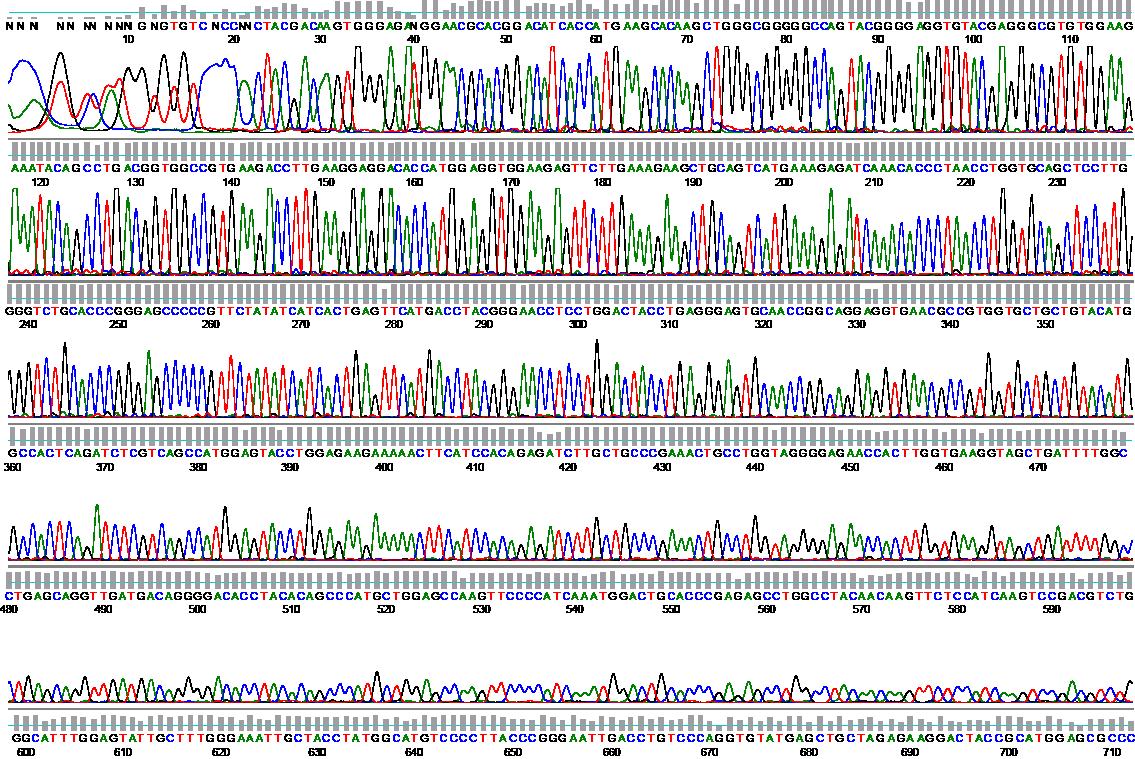
Beispiel: BCR-ABL-Mutationsanalyse mittels Sequenzierung
Präanalytische Hinweise
Antikoagulans für alle molekulargenetischen Untersuchungen ist grundsätzlich EDTA. In Ausnahmefällen geht auch Zitrat. Heparin ist dagegen ungünstig (hemmt enzymatische Reaktionen wie die PCR) und sollte nur dann verwendet werden wenn nichts anderes verfügbar ist (z. B. versehentliche Abnahme in einer falschen Spritze bei einer Knochenmarkpunktion).
Als Untersuchungsmaterial sind im Prinzip möglich:
- Knochenmarkaspirat (5 ml)
- Peripheres Blut (10 ml)
- Erguss-Material (Aszites, Pleuraerguss)
- Liquor
- Lymphknoten- oder Tumorbiopsie (unbedingt unfixiert, d.h. in NaCl oder Wasser !)
Bei Einsendung von Tumormaterial oder Lymphknoten wird um vorherige telefonische Rücksprache gebeten. Die molekulargenetische Diagnostik aus Liquor hat meist nur dann Aussicht auf Erfolg, wenn dieser sehr zellreich ist. Auch hier wird eine vorherige telefonische Rücksprache empfohlen.
Bei Chimärismusanalysen nach allogener Transplantation muss sichergestellt sein, dass unserem Labor Material sowohl vom Spender, als auch vom Empfänger vor Transplantation vorliegt bzw. vorlag. Voraussetzung für eine Chimärismusanalyse ist die Kenntnis der Unterschiede in den Chimärismusmarkern zwischen Spender und Empfänger. Die Chimärismusanalyse kann entweder „unsortiert“ („Gesamtchimärismus“) aus Gesamt-Blut, – Knochenmark, etc., oder „sortiert“ („sortierter Chimärismus“), d. h. aus isolierten Zellfraktionen, z. B. CD34+ Stammzellen, erfolgen. Falls ein „sortierter Chimärismus“ angefordert wird, muss das Material frisch sein, d. h. die auszusortierenden Zellen müssen noch intakt sein.
Der Probenversand sollte insbesondere bei zeitkritischen Proben (akute Leukämie) per Eilpost erfolgen. Der Versand sollte wenn möglich nicht an einem Freitag erfolgen.
-
In der Tumorzytogenetik werden erworbene Chromosomenanomalien bei hämatologischen Neoplasien und soliden Tumoren untersucht. Charakteristische Chromosomenanomalien, die häufig bei einem bestimmten Tumortyp auftreten, werden mit der neoplastischen Transformation assoziiert: Sie gelten als primäre Anomalien. Sekundäre Anomalien treten beim Fortschreiten der Erkrankung auf, wenn das Genom zunehmend instabil wird. Sie können zur Progression des Tumors beitragen.
Was kann die Tumorzytogenetik leisten?
Die zytogenetische Untersuchung von peripherem Blut und Knochenmark gehört heute zur Standarddiagnostik maligner hämatologischer Erkrankungen. Sie umfasst die klassische Chromosomenanalyse zusammen mit der Fluoreszenz-in-situ-Hybridisierung. Die zytogenetische Analyse leistet einen wichtigen Beitrag, die Verdachtsdiagnose, die Prognoseabschätzung, die Therapie-Optionen zu sichern und zu spezifizieren. Außerdem dient sie zur Kontrolle des Krankheitsverlaufs. Die Identifizierung spezifischer, genetischer Marker des malignen Zellklons kann außerdem dazu herangezogen werden, den Therapie-Verlauf zu überwachen.Indikationen
Myeloische Leukämien
- Myeloproliferative Neoplasien (MPN)
- Myelodysplastische / myeloproliferative Erkrankungen
- Myelodysplastische Syndrome (MDS)
- Akute myeloische Leukämien (AML)
- Biphänotypische akute Leukämien (BAL)
Akute lymphatische Leukämien
- B-Zell akute lymphatische Leukämien (B-ALL)
- T-Zell akute lymphatische Leukämien (T-ALL)
- Non-Hodgkin-Lymphome (NHL)
Methoden
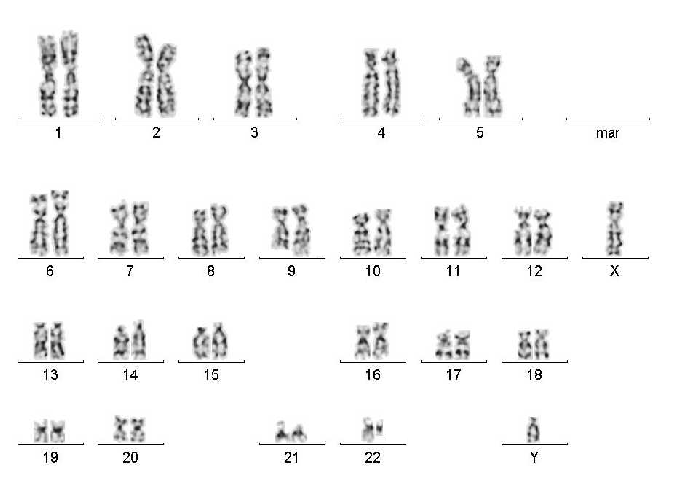
Karyogramm
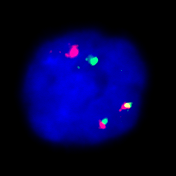
Fluoreszenz-in-situ-Hybridisierung
Eine Chromosomenanalyse erfolgt an präparierten Metaphasen: mittels computergestützter Karyotypisierung.
Mit der FISH-Analyse lassen sich noch feinere submikroskopische Veränderungen sichtbar machen. Sie ermöglicht auch ein Screening auf häufige, tumortypische Aberrationen bei ungenügendem Proliferationsverhalten der Tumorzellen (etwa Plasmozytom oder Chronisiche lymphatische Leukämie).
Die Spektral-Karyotypisierung (SKY) stellt eine wichtige Weiterentwicklung der FISH-Methode dar. In der molekularzytogenetischen Tumordiagnostik gewinnt sie zunehmend an Bedeutung. Mittels SKY können auch komplexe aberrante Rearrangements mit unbekannten Translokationspartnern oder Markerchromosomen unbekannter Herkunft aufgeschlüsselt werden.
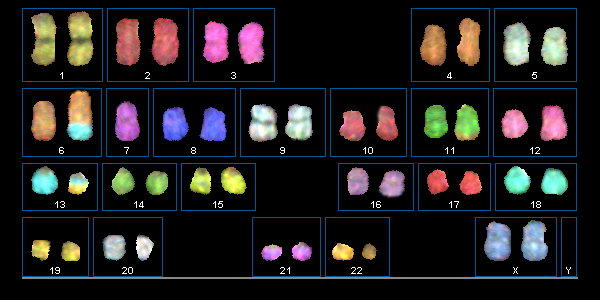
Spektral-Karyotypisierung
Präanalytische Hinweise
Na-Heparin Knochenmark 10ml / Peripheres Blut 20ml
Krankheitsentitäten
Die untergeordneten Seiten enthalten einige allgemeine Informationen und diagnostische Hinweise zu verschiedenen hämatologischen Erkrankungen. Die Einteilung in Krankheitsgruppen orientiert sich dabei an der WHO-Klassifikation der hämatologischen Neoplasien aus dem Jahr 2008.
Jedes Unterkapitel enthält Hinweise, wie eine möglichst rationale Diagnostik durchzuführen ist.
Beispielsweise werden in den folgenden Kapiteln Hinweise gegeben, welche genetischen Untersuchungen wann sinnvoll sind (meist in Abhängigkeit von den zugehörigen immunologischen und morphologischen Befunden).
- Akute myeloische Leukämie
- Akute lymphatische Leukämie
- Myelodysplastische Syndrome
- Eosinophilen-Syndrome und
- Non-Hodgkin-Lymphome
In folgenden Kapiteln wird auch auf ausgewählte einzelne Erkrankungen detaillierter eingegangen.
- Non-Hodgkin-Lymphome
- Myelodysplastische Syndrome
- MDS/MPN-Mischformen und
- Myeloproliferative Neoplasien
-
Die laborchemische Basisdiagnostik bei MPN umfasst das mikroskopische Differenzialblutbild, die Bestimmung klinisch-chemischer Parameter und die Knochenmarkdiagnostik.
Die Knochenmarkdiagnostik sollte bei Erstdiagnose die folgenden Untersuchungen umfassen:
- Knochenmark-Histologie
- Knochenmark-Zytologie
- Immunphänotypisierung
- Molekulargenetik
- Zytogenetik
Die Knochenmark-Histologie ist dabei die einzige der o. g. Untersuchungen, die nicht im Labor Berlin angeboten wird. Sie erlaubt Aussagen über den Fibrosegrad im Knochenmark und ist von Bedeutung bei der Differenzialdiagnose Myeloproliferativer Neoplasien.
Chronische Myeloische Leukämie (CML)
In der Knochenmarkzytologie sieht man bei der chronischen myeloischen Leukämie meist eine Steigerung aller 3 Zellreihen (Megakaryopoese, Myelopoese, Erythropoese), wobei die Myelopoese deutlich dominiert, oft verbunden mit einer Eosinophilie und Basophilie.
Molekulargenetisch ist fast immer das Fusionsgen BCR-ABL mittels RT-PCR nachweisbar. Wenn die molekulargenetische Analyse dieses Fusionsgen nicht nachweisen kann ist eine CML unwahrscheinlich. Konventionell-zytogenetisch lässt sich gelegentlich keine Translokation t(9;22) bzw. kein Philadelphia-Chromosom detektieren, jedoch molekulargenetisch die BCR-ABL-Fusion. Die molekulargenetische Analyse kann bei V. a. CML auch aus dem peripheren Blut erfolgen. Da es viele verschiedene Transkriptvarianten von BCR-ABL gibt muss die molekulargenetische Analyse darauf ausgerichtet sein alle diese nachweisen zu können.Die zytogenetische Analyse sollte aus dem Knochenmark und nur wenn nicht anders möglich aus dem peripheren Blut durchgeführt werden, da sich die aus dem peripheren Blut gewonnenen Zellen in Kultur nicht selten nur unzureichend zur Teilung anregen lassen. Zytogenetisch sieht man bei CML typischerweise die Translokation t(9;22)(q34;q22), die oft ein verkürztes Chromosom 22 („Philadelphia-Chromosom“) zeigt.
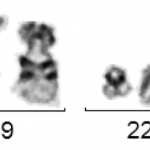
t(9;22)(q34;q11) mit Philadelphia-Chromosom
Der Wert der zytogenetischen Analyse liegt aber nicht nur im Nachweis der t(9;22), sondern auch darin, dass andere zusätzliche Aberrationen nachgewiesen werden können. Typische Zusatzaberrationen bei CML sind der Verlust des langen Arms von Chromosom 9 (del(9q)), ein zusätzliches Philadelphia-Chromosom bzw. eine zweite t(9;22) sowie eine Inversion von Chromosom 17 (inv(17)). Alle drei Zusatzaberrationen gelten als prognostisch ungünstig, werden häufiger in der akzelerierten Phase oder dem Blastenschub gefunden und können ein Hinweis auf rasche Progredienz der Erkrankung sein. Außerdem gibt es sehr selten Fälle, in denen klinisch das Bild einer CML, aber genetisch keine Translokation t(9;22) oder kein Fusionsgen BCR-ABL, sondern eine Fusion des BCR-Gens mit anderen Tyrosinkinasen (z. B. BCR-FGFR3, BCR-JAK2, etc.) vorliegt. Die Zytogenetik sollte immer durch eine Fluoreszenz-in-situ-Hybridisierung (FISH) für das Fusionsgen BCR-ABL ergänzt werden.
Verlaufsuntersuchungen bei CML
Verlaufsuntersuchungen bei BCR-ABL-positiver CML werden mittels quantitativer PCR (qPCR) durchgeführt. Bei einem hohen Krankheitsniveau sind auch zusätzliche zytogenetische (+FISH) Untersuchungen sinnvoll. Als Untersuchungsmaterial ist für die Molekulargenetik peripheres Blut ausreichend, da sich gezeigt hat, dass das BCR-ABL-Niveau zwischen Knochenmark und Blut relativ gut korreliert. Durch das European Leukemia Net wurden bestimmte zeitliche Landmarken definiert, die bei einer Therapie mit Tyrosinkinase-Inhibitoren erreicht werden sollen. Werden diese Zielmarken nicht erreicht, kann dies als Warnsignal gelten. Bei Patienten unter Therapie mit Tyrosinkinase-Inhibitoren sollte alle 3 Monate eine qPCR aus peripherem Blut durchgeführt werden.BCR-ABL-Mutationsanalysen
Falls es unter der Therapie mit einem Tyrosinkinase-Inhibitor bei mehreren zeitlich aufeinanderfolgenden qPCR-Untersuchungen zu einem kontinuierlichen deutlichen Anstieg des BCR-ABL-Niveaus kommt (um mehr als eine Größenordnung), sollte eine BCR-ABL-Mutationsanalyse durchgeführt werden. Diese erfolgt mittels direkter Sequenzierung aus dem zuletzt eingesandten Material mit dem höchsten BCR-ABL-Niveau. Falls eine BCR-ABL-Mutation gefunden wird, kann dies unter Umständen eine wertvolle Entscheidungshilfe für den Wechsel auf einen anderen Tyrosinkinaseinhibitor sein.BCR-ABL-negative Myeloproliferative Neoplasien (MPN)
Die Mehrheit der MPN weisen nicht das Fusionsgen BCR-ABL auf. Oft lassen sich aber andere molekulare Aberrationen nachweisen.
MPN mit JAK2 V617F-Mutation
Seit der Erstbeschreibung im Jahr 2005 ist bekannt, dass viele MPN molekular durch eine Punktmutation in der Januskinase 2 (JAK2) gekennzeichnet sind. Diese Punktmutation führt zum Aminosäureaustausch Valin -> Phenylalanin an Position 617 des JAK2-Proteins (V617F-Mutation) und konsekutiv zur konstitutionellen Aktivierung der JAK2-Kinase.Die JAK2 V617F-Mutation ist in unterschiedlichem Prozentsatz bei verschiedenen MPN zu finden – in mehr als 90% bei Polyzythämia vera (PV), und in einem deutlich geringeren Prozentsatz bei Primärer Myelofibrose (PMF) oder Essentieller Thrombozythämie (ET). Ein negativer JAK2 V617F-Befund schließt also die genannten Erkrankungen nicht aus. Die JAK2-Mutationen wurde durch die WHO als Majorkriterium für die Diagnosestellung einer PV, PMF oder ET aufgenommen.
MPN mit JAK2 Exon 12-Mutationen
Wesentlich seltener als die JAK2 V617F-Mutation, der eine Nukleotidmutation im JAK2 Exon 14 zugrundeliegt, finden sich Mutationen in anderen Abschnitten des JAK2-Gens. Vor allem sind diese in Exon 12 des JAK2-Gens zu finden. Patienten mit JAK2 Exon 12-Mutationen weisen fast immer das Bild einer isolierten Erythrozytose, d.h. einer Polyzythämia vera auf. Diese Mutationen sind sehr selten und sollten nicht routinemäßig bei jeder MPN-Diagnostik mitbestimmt werden. Zu empfehlen ist die Diagnostik bei klinischem Bild einer PV und gleichzeitig nicht nachweisbarer JAK2 V617F-Mutation.MPN mit Calreticulin (CALR)-Mutation
Seit dem Jahr 2013 ist bekannt, dass etwa 25 bis 30 % der Fälle von PMF und ET Mutationen im letzten codierenden Exon des Gens CALR aufweisen. Bei PV kommen diese Mutationen so gut wie nie vor. Die Mutationen führen zu einer Leserahmenverschiebung um -1 Nukleotid, mit der Folge, dass das transkribierte mutierte Calreticulin-Protein ein verändertes carboxyterminales Ende aufweist. Dadurch kommt es zu einer Fehlsortierung des Proteins in subzelluläre Kompartimente. Hauptsächlich werden zwei rekurrente Mutationstypen beobachtet – „Typ 1“ und „Typ 2“, aber ist gibt auch zahlreiche weitere. CALR-Mutationen gelten grosso modo als prognostisch günstiger als JAK2-Mutationen.MPN mit MPL W515L-Mutation
Bei ET und PMF findet sich gelegentlich eine Punktmutation im Thrombopoetin-Rezeptorgen MPL, die zum Aminosäureaustausch Tryptophan gegen Leucin an Position 515 (W515L) führt. Die Mutation findet sich in 5% oder weniger aller Patienten mit ET oder PMF, und zwar häufiger bei PMF als bei ET. Der Nachweis der MPL W515L-Mutation gehört zu den WHO-Diagnose-Majorkriterien der PMF und ET.Systemische Mastozytose
Die systemische Mastozytose weist bei erwachsenen Patienten häufig aktivierende Mutationen in der auf Chromosom 4q11 gelegenen Tyrosinkinase KIT auf. Am weitaus häufigsten wird dabei der Aminosäureaustausch Aspartat -> Valin an Position 816 (D816V) gefunden.Diagnosekriterien der WHO für PV, ET und PMF aus dem Jahr 2008
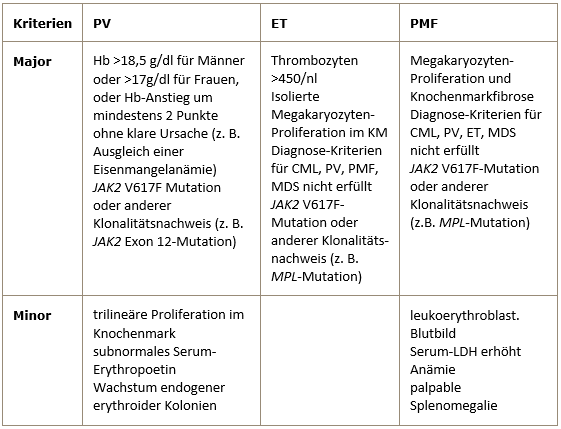
Die Diagnosestellung der PV erfordert entweder beide Majorkriterien und ein Minorkriterium oder das erste Majorkriterium und zwei Minorkriterien.
Die Diagnosestellung der ET erfordert alle 4 Majorkriterien.
Die Diagnosestellung der PMF erfordert alle 3 Majorkriterien und zwei Minorkriterien. -
Unter dem Begriff der Myelodysplastischen Syndrome (MDS) wird eine heterogene Gruppe von klonalen myeloischen Erkrankungen zusammengefasst. Gemeinsames Merkmal dieser Erkrankungen sind die Ausreifungsstörungen der Myelopoese im Knochenmark, verbunden mit einer peripheren Zytopenie. Die MDS müssen zum einen differenzialdiagnostisch zur akuten myeloischen Leukämie (AML) und zum anderen zu den Myeloproliferativen Neoplasien (MPN) abgegrenzt werden. Zur Basisdiagnostik bei Verdacht auf MDS gehören:
- Knochenmark-Zytologie
- Zytogenetik des Knochenmarks
- Mikroskopisches Differenzialblutbild
- Knochenmark-Histologie
- Immunphänotypisierung (Durchflusszytometrie) des Knochenmarks
- Molekulargenetik
Die beiden wichtigsten Knochenmark-Untersuchungen bei der Diagnostik der MDS sind die Morphologie und die zytogenetische Analyse. Die Knochenmark-Histologie sollte bei einem Hämatopathologen analysiert werden.
Historisch wurden die MDS auch unter dem Begriff „Präleukämie“ geführt. Dieser Begriff rührt daher, dass alle MDS das Risiko in sich tragen, in eine akute myeloische Leukämie (AML) überzugehen. Dieses Risiko ist je nach MDS-Typ jedoch sehr unterschiedlich. Der Übergang vom MDS in die AML ist fließend und die Differenzialdiagnose MDS<->AML muss letztlich auch klinisch gestellt werden. Ab 20% Blasten im Knochenmark spricht man definitionsgemäß von einer AML, bei unter 20% liegt ein MDS vor. Zur Primärdiagnostik gehört auch das mikroskopische Differenzialblutbild. Hier kommt die Art und Zahl der Zytopenien zum Ausdruck (wichtig für die MDS-Klassifikation), zum anderen muss auch die Zahl der Monozyten beachtet werden. Bei einer persistierenden Monozytose >1000/µl liegt definitionsgemäß eine chronische myelomonozytäre Leukämie (CMML) vor, die zu den MDS/MPN-Mischformen gezählt wird.
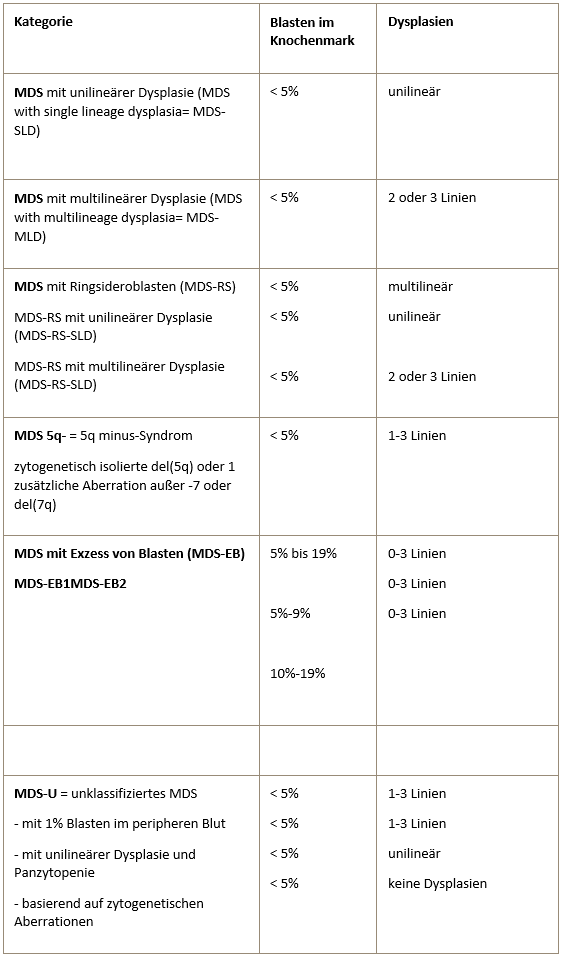
WHO-Klassifikation (2016) der Myelodysplastischen Syndrome
Knochenmarkzytologie
Die Knochenmarkzytologie ist von großer Bedeutung bei der Diagnosestellung und Verlaufsbeurteilung eines MDS. Zum einen wird hiermit der Blastenanteil und zum anderen werden die dysplastischen Veränderungen in den verschiedenen Zellreihen beurteilt. Außerdem wird damit das Vorhandensein von Ringsideroblasten erfasst. Ringsideroblasten sind Erythroblasten, die in der Berliner-Blau-Färbung perinukleär angeordnete Granula (sogenannte Siderosomen) aufweisen. Bei diesen Granula handelt es sich um mit Ferritin beladene Mitochondrien. Ringsideroblasten kommen in geringer Zahl auch im gesunden Knochenmark vor. Beim MDS sind sie Ausdruck einer Eisenverwertungsstörung.
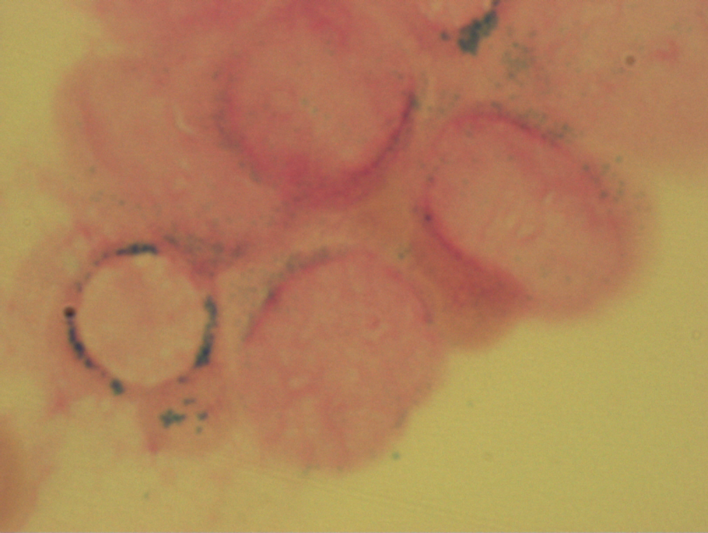
Ringsideroblasten bei RARS
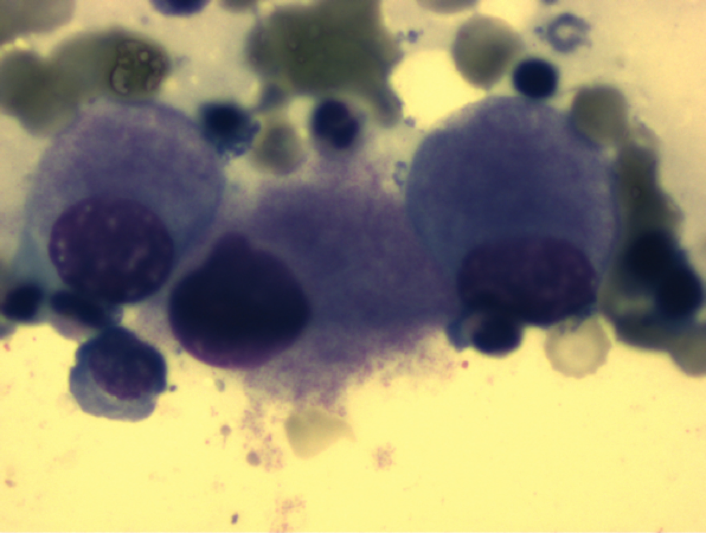
Reifungsgestörte Megakaryozyten bei MDS 5q-
Durchflusszytometrie
Die Immunphänotypisierung mittels Durchflusszytometrie sollte ergänzend durchgeführt werden, auch um den Anteil myeloischer Blasten abzuschätzen.
Genetik
Die genetische Grundlage der Myelodysplastischen Syndrome ist heterogen. Eine Vielzahl von genetischen Veränderungen, denen zum Teil auch prognostische Bedeutung zukommt und die auch variabel miteinander kombiniert auftreten, sind bekannt. Bisher haben vor allem die zytogenetisch definierten Veränderungen Bedeutung, da hier klare Risikogruppen identifiziert werden konnten. Daher hat die zytogenetische Analyse beim MDS einen ähnlich hohen Stellenwert wie die Knochenmark-Morphologie. Bei jeder Abklärung eines MDS-Verdachts sollte eine gründliche zytogenetische Analyse durchgeführt werden. Die Zytogenetik des Knochenmarks sollte auch eine Fluoreszenz in situ-Hybridisierung (FISH) für die wichtigsten bei MDS vorkommenden zytogenetischen Aberrationen umfassen.
Prognostische Wertigkeit häufiger zytogenetischer Aberrationen bei MDS

Alle nicht in dieser Tabelle enthaltenen Aberrationen werden der intermediären prognostischen Risikogruppe zugeordnet.
Das Panel am FISH-Sonden für die MDS-Diagnostik umfasst im Labor Berlin die folgenden Sonden/Genloci:
- 5q31/5q33 (5q-Deletion, Monosomie 5)
- 7q31 (7q31-Deletion, Monosomie 7)
- 20q12 (20q12-Deletion, Monosomie 20)
- CEP 8 (Trisomie 8)
- TP53 (17p13-Deletion)
- CEN Y (Verlust des Y-Chromosoms)
- TEL (ETV6) (12p13-Deletion)
5q minus-Syndrom
Die häufigste zytogenetische Aberration bei MDS sind Deletionen des langen Arms von Chromosom 5 (del(5q)). Eine „minimal deletierte Region“ wurde im Bereich 5q31-32 lokalisiert.
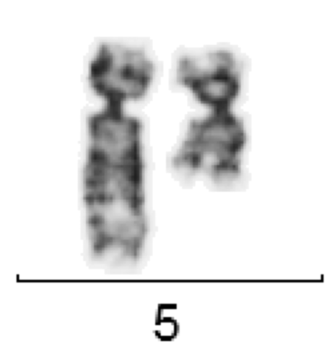
del(5)(q12Q33) bei MDS
Von den im Bereich dieser Region liegenden Genen ist bisher weitgehend unbekannt, welche Rolle sie bei der molekularen Pathogenese der MDS spielen. Das 5q minus-Syndrom (MDS 5q) ist durch eine partielle oder vollständige del(5q) als einzige zytogenetische Aberration (d.h. ohne weitere Zusatzaberrationen) oder mit einer zusätzlichen Aberration (außer -7 oder del(7q) gekennzeichnet.Der Blastenanteil im KM liegt unter 5%. Typischerweise finden sich bei MDS 5q- im Knochenmark ungewöhnlich kleine Megakaryozyten mit monolobuliertem Kern.
Molekulargenetik
In den letzten Jahren wurden viele molekulargenetische Aberrationen bei MDS beschrieben. Zu den betroffenen Genen gehören Spleißfaktoren, wie SRSF2 oder SF3B1, Tumosuppressoren wie TP53, epigenetische Regulatoren wie TET2, DNMT3A oder ASXL1, Transkriptionsfaktoren, wie RUNX1, und andere. Jede dieser Gen-Aberrationen liegt jedoch in weniger als 10-20% der MDS-Fälle vor. Für einige Gene sind relativ klare ungünstige prognostische Implikationen bekannt (TP53, ASXL1, EZH2). Die meisten dieser Aberrationen sind allerdings nicht MDS-spezifisch und finden sich auch bei anderen myeloischen Neoplasien (AML, MPN). AML-typische Aberrationen wie t(15;17)/PML-RARA, inv(16)/CBFB-MYH11, t(8;21)/AML1-ETO kommen bei echtem MDS nicht vor. Molekulargenetische Untersuchungen (typischerweise als „Gen-Panel“) bei MDS können zur Diagnosesicherung, zur Prognoseabschätzung, zur Gewinnung molekularer Marker für eventuelle spätere quantitative Verlaufsbeobachtungen und auch zur Identifizierung von Zielgenen/proteinen für eine eventuelle gezielte Therapie dienen.
-
MDS-MPN-Mischformen
Es gibt maligne hämatologische Erkrankungen, die sowohl Merkmale einer Myeloproliferation (im peripheren Blutbild sichtbare Steigerung von mindestens einer Zellreihe), als auch Merkmale einer Myelodysplasie (dysplastisch veränderte Myelopoese im Knochenmark) tragen. In der WHO-Klassifikation 2008 wurde daher die Entität „Myelodysplastische / Myeloproliferative Neoplasien“ geschaffen. Diese Entität muss gegenüber den MPN und MDS differenzialdiagnostisch abgegrenzt werden. Die Knochenmarkdiagnostik entspricht der bei MPN und MDS durchzuführenden. Zwei etwas genauer definierte MDS/MPN-Formen werden beispielhaft kurz beschrieben.
Chronische myelomonozytäre Leukämie (CMML)
Die CMML ist durch die folgenden Merkmale definiert:
- über längere Zeit persistierende Monozytose > 1000/µl im peripheren Blut ohne andere erkennbare Ursache
- Dysplasien im einer oder mehreren Reihen der Hämatopoese im Knochenmark
- kein Nachweis von BCR-ABL oder eines Philadelphia-Chromosoms
- Blastenanteil im Knochenmark < 20%
Der genetische Hintergrund der CMML ist nicht einheitlich und bisher nicht eindeutig entschlüsselt. In einer kleinen Minderheit der Fälle (unter 5 %) ist die JAK2 V617F-Mutation nachweisbar. Von besonderer diagnostischer Bedeutung sind die Knochenmark-Zytologie und die zytogenetische Untersuchung.
Refraktäre Anämie mit Ringsideroblasten und Thrombozytose (RARS-T)
Die RARS-T ist definiert durch die folgenden Merkmale:
- < 5 % Blasten im Knochenmark
- Dysplasien in einer oder mehreren Zellreihen im Knochenmark
- > 15 % Ringsideroblasten im Knochenmark
- Thrombozytose > 600/nl im peripheren Blut
Die wichtigsten Differenzialdiagnosen sind die Essentielle Thrombozythämie (ET; dort finden sich keine höhergradigen Dysplasien und keine Vermehrung von Ringsideroblasten) und das RARS bzw. RCMD (diese zeigen keine Thrombozytose).
In der Mehrheit der Fälle lässt sich bei RARS-T die JAK2 V617F-Mutation nachweisen.
-
Einige maligne hämatologische Erkrankungen sind mit einer Eosinophilie verbunden. Besonders charakteristisch ist dies für die Myeloproliferativen Neoplasien. Aber auch beispielsweise bei manchen T-Zell-Lymphomen findet sich eine Eosinophilie.
Zur Basisdiagnostik bei V. a. maligne Erkrankung mit Eosinophilie gehören
- Knochenmark-Histologie
- Knochenmark-Zytologie
- Immunphänotypisierung
- Molekulargenetik
- Zytogenetik.
Die genetische Basis von malignen Eosinophilen-Erkrankungen ist heterogen und bisher nur zum Teil bekannt. In einem Teil der Fälle liegen Aberrationen bestimmter Gene vor:
- PDGFRA (platelet-derived growth factor receptor alpha, alpha-Rezeptor für Thrombozyten-Wachstumsfaktor) auf Chromosom 4q12.
- PDGFRB (platelet-derived growth factor receptor beta, beta-Rezeptor für Thrombozyten-Wachstumsfaktor) auf Chromosom 5q33.1.
- FGFR1 (fibroblast growth factor receptor 1, Rezeptor für den Fibroblastenwachstumsfaktor 1) auf Chromosom 8p12.
Alle 3 Gene sind Tyrosinkinasen. Vermutlich sind auch noch weitere bisher nicht identifizierte Tyrosinkinasen an der Ätiopathogenese maligner Eosinophilenerkrankungen beteiligt.
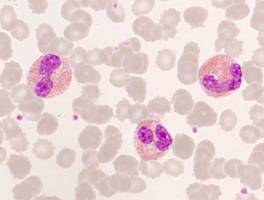
Eosinophile im peripheren Blut
Von entscheidender Bedeutung ist eine genaue zytogenetische Analyse (möglichst immer aus Knochenmark). Diese zytogenetische Analyse sollte eine Fluoreszenz-in situ-Hybridisierung (FISH) für die Gene PDGFRA, PDGFRB und FGFR1 einschließen.
Bei einem kleinen Teil der Fälle von chronischer Eosinophilenleukämie findet sich ein Fusionsgen FIP1L1-PDGFRA. Dieses Fusionsgen kann molekulargenetisch mittels RT-PCR oder per FISH-Analyse nachgewiesen werden. Es kommt durch eine Mikrodeletion auf Chromosom 4q12 zustande, die konventionell-zytogenetisch nicht sichtbar ist und bei der die benachbart liegenden Gene FIP1L1 und PDGFRA miteinander fusioniert werden.
Der Nachweis von PDGFRA- oder PDGFRB-Aberrationen hat unmittelbare therapeutische Konsequenzen: beide Tyrosinkinasen lassen sich sehr gut mit Imatinib, Nilotinib oder Dasatinib hemmen.
Mastzellerkrankungen
In der WHO-Klassifikation der hämatologischen Neoplasien 2017 wurde die Mastozytose als separate Krankheitsentität definiert (zuvor war sie unter die Myeloproliferativen Neoplasien eingeordnet worden). Unterschieden werden die folgenden Formen:
- Kutane Mastozytose
- Indolente Systemische Mastozytose
- Systemische Mastozytose mit assoziierter hämatologischer Neoplasie (SM-AHN)
- Aggressive Systemische Mastozytose (ASM)
- Mastzellleukämie
- Mastzellsarkom
In der großen Mehrheit der aller Mastozytose-Formen finden sich aktivierende somatische Mutationen in der Tyrosinkinase KIT. Bei weitem am häufigsten ist die KIT D816V-Mutation, die sich mit hoher Sensitivität mittels quantitativer PCR auch im Blut nachweisen lässt, selbst bei klinischen Formen, die scheinbar keine hämatogene Aussaat zeigen. Falls die KIT D816V-Mutation nicht nachweisbar ist, aber weiterhin der hochgradige Verdacht auf eine klonale Mastzellerkrankung besteht, sollte eine KIT-Sequenzierung erfolgen, bei der auch andere, seltenere KIT-Mutationen erfasst werden können. In Abhängigkeit von KIT-Mutationstyp können verschiedene Tyrosinkinaseinhibitoren zum Einsatz kommen.
-
Die Knochenmarkdiagnostik bei AML umfasst:
- Knochenmark-Zytologie
- Immunphänotypisierung
- Molekulargenetik
- Zytogenetik.
Die Knochenmark-Histologie ist in Regel entbehrlich und nur in Sonderfällen (punctio sicca) erforderlich.
Zytomorphologie
Die Zytomorphologie besitzt bei der AML eine größere Bedeutung als bei der ALL, da sich manche Subtypen der AML schon zytomorphologisch diagnostizieren lassen. Häufig wird die AML noch nach der FAB (French-American-British)-Klassifikation eingeteilt. Diese Klassifikation geht in ihren Anfängen auf das Jahr 1976 zurück und ist eine rein zytomorphologische Einteilung. Modernere Klassifikationen, wie die WHO-Klassifikation aus dem Jahr 2016 berücksichtigen dagegen auch genetische und immunologische Aspekte. Besondere Bedeutung hat die Zytomorphologie bei der Erkennung des FAB-M3-Subtyps (akute Promyelozytenleukämie). Auch der FAB-M4Eo-Subtyp (akute myelomonozytäre Leukämie mit abnormen Eosinophilen) lässt sich in der Regel schon zytomorphologisch erkennen.
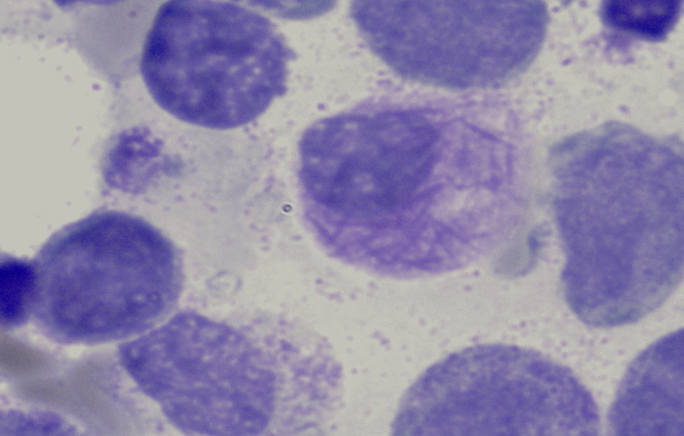
Bündel von Auerstäbchen bei AML M3
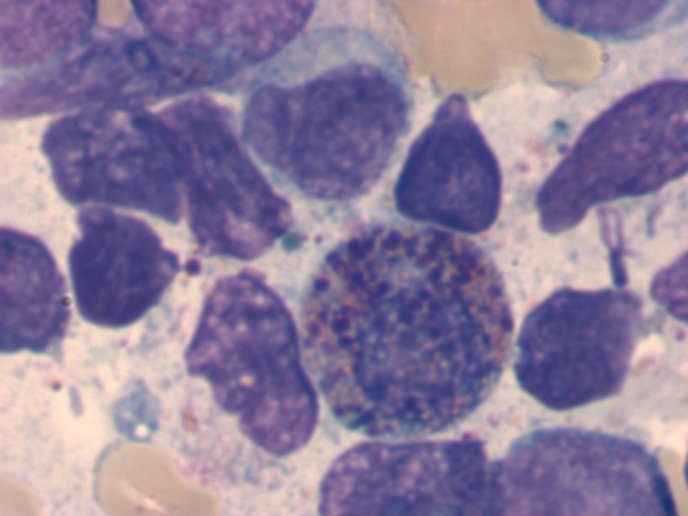
Abnormer Eosinophiler bei AML M4Eo
Durchflusszytometrie
Durch Immunphänotypisierung mittels Durchflusszytometrie lässt sich die Zugehörigkeit einer akuten Leukämie zur myeloischen Reihe sichern. Allerdings hat die Durchflusszytometrie bei der weiteren Subtypisierung in der diagnostischen Routine bei AML einen geringeren Stellenwert als bei der ALL, da sich die einzelnen AML-Subtypen (FAB oder WHO) immunzytologisch nicht mit ausreichender Sicherheit voneinander abgrenzen lassen.
Zytogenetik und Molekulargenetik
Die Zyto- und insbesondere die Molekulargenetik gewinnt für die AML immer größere Bedeutung. Mittlerweile ist eine große Zahl von genetischen Aberrationen beschrieben. Das Bild wird dadurch noch unübersichtlicher, weil diese genetischen Aberrationen häufig miteinander kombiniert auftreten können.
Die genetische Basisdiagnostik bei jeder AML sollte in jedem Fall eine konventionelle zytogenetische Analyse umfassen. In der folgenden Tabelle sind die Indikationen für molekulargenetische Untersuchungen aufgeführt.
Indikationen für molekulargenetische Untersuchungen
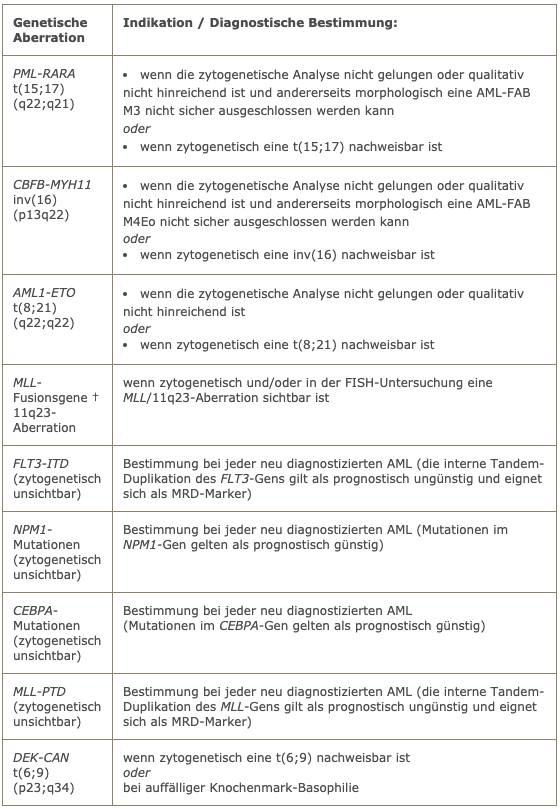

† die bei AML häufigsten MLL-Fusionsgene sind:
- MLL-AF9/t(9;11)(p22;q23)
- MLL-ENL/t(11;19)(q23;p13.3)
- MLL-ELL/t(11;19(q23;p13.1)
- MLL-AF6/t(6;11)(q27;q23)
- MLL-AF10/t(10;11)(p13;q23)
Zusammen machen sie mehr als 80% aller MLL-Translokationen bei AML aus.
-
Die Knochenmarkdiagnostik bei ALL umfasst:
- Knochenmark-Zytologie
- Immunphänotypisierung
- Molekulargenetik
- Zytogenetik.
Die Knochenmark-Histologie ist in Regel entbehrlich und nur in Sonderfällen (punctio sicca) erforderlich.
Etwa ein gutes Drittel aller Fälle von ALL tritt im Kindesalter (< 15 J.) auf, der Rest im Adoleszenten- und Erwachsenenalter (≥ 15 J.). Die genaue immunologische und genetische Charakterisierung dient zum einen der Diagnosesicherung, als auch der Risikostratifizierung, da sich gezeigt hat dass verschiedene ALL-Subtypen eine ganz unterschiedliche Prognose haben. Außerdem eröffnet die genetisch-immunologische Charakterisierung Möglichkeiten zu einer zielgerichteten Therapie (z. B. Imatinib, Rituximab, Blinatumomab).
Durchflusszytometrie
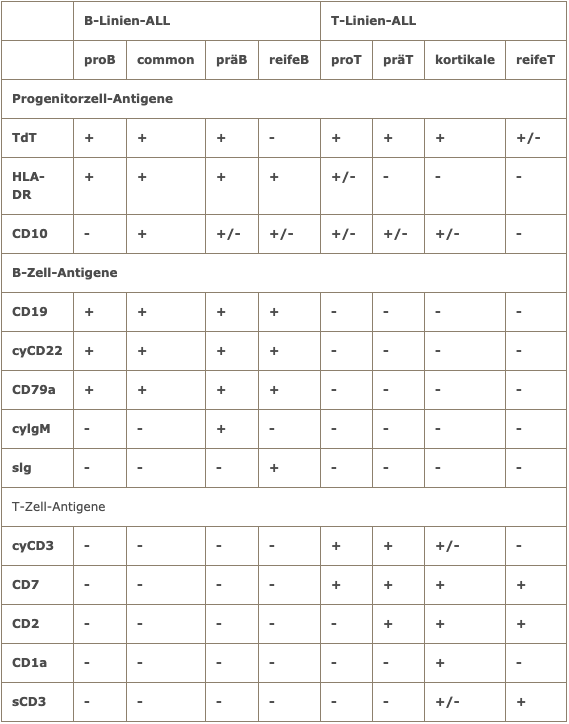
Die durchflusszytometrische Analyse ist bei ALL von zentraler Bedeutung. Sie erlaubt die Unterscheidung von B-Linien- und T-Linien-ALL. Innerhalb der B-Linien-ALL ist die Abgrenzung der reifen B-ALL von der B-Vorläufer-ALL von großer therapeutischer Relevanz.
Immunologische Einteilung der ALL nach EGIL (European Group for the immunological classification of acute leukemias)
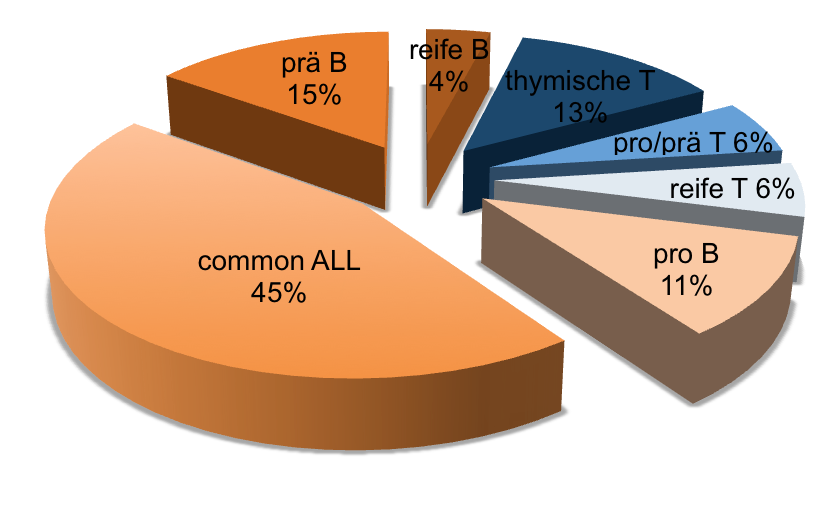
Ungefähre Häufigkeit verschiedener Immunphänotypen bei Erwachsenen-ALL
Untersuchungsmaterial für die Durchflusszytometrie ist Knochenmarkaspirat oder blastenreiches peripheres Blut. In Sonderfällen können auch (unfixierte) Lymphknoten untersucht werden.
Genetik
Der genetische Hintergrund der ALL im Kindesalter und im Erwachsenen- und Adoleszentenalter unterscheidet sich erheblich. Viele genetische Aberrationen bei ALL zeigen eine ausgesprochene Altersabhängigkeit. Dementsprechend werden die diagnostischen Akzente bei Kindern und Erwachsenen meist etwas anders gesetzt.
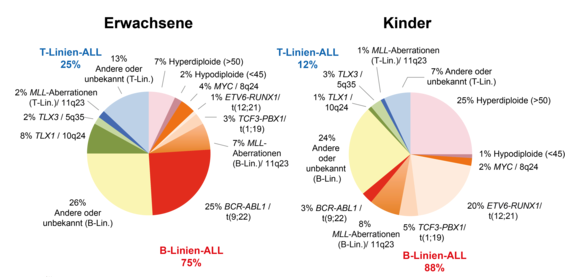
Ungefähre Verteilung genetischer Aberrationen bei ALL
Die bei ALL zu findenden genetischen Aberrationen sind häufig eng mit bestimmten Immunphänotypen korreliert.
Häufige genetische Aberrationen bei ALL
Zytogenetik
Die zytogenetische Untersuchung ist obligat. Sie sollte bei B-Vorläufer-ALL auch eine FISH-Untersuchung auf BCR-ABL sowie eine FISH-Untersuchung auf MLL-Aberration umfassen. Bei reifer B-ALL sollte eine FISH-Untersuchung auf MYC-Translokation durchgeführt werden.
Molekulargenetik
Das molekulargenetische Untersuchungsprogramm orientiert sich am Immunphänotyp. Bei B-Vorläufer-ALL ist eine Untersuchung auf BCR-ABL obligat. Bei CD10-negativer ALL sollten die beiden häufigsten MLL-Fusionsgene, d.h. MLL-AF4 und MLL-ENL untersucht werden. Bei prä B-ALL sollte eine Untersuchung auf E2A-PBX1 erfolgen. Bei reifer B-ALL sollte die MYC-IGH-Fusion untersucht werden. Bei T-Linien-ALL ist eine Untersuchung auf NUP214-ABL sinnvoll.
-
Die Non-Hodgkin-Lymphome bilden eine heterogene Gruppe von Erkrankungen. Zur Basisdiagnostik gehören:
- Lymphknoten- und/oder Knochenmark-Histologie
- Immunphänotypisierung (Durchflusszytometrie)
- Zytogenetik
- Molekulargenetik
- Knochenmark-Zytologie
Besondere Bedeutung kommt in der Regel der Lymphknoten- oder Knochenmark-Histologie zu, die bei einem Pathologen analysiert wird. In schwierigen Fällen sollte immer ein Referenzpathologe für Lymphomerkrankungen hinzugezogen werden. Die Knochenmarkdiagnostik dient ansonsten zum einen der Diagnosesicherung, zum anderen auch der Ausbreitungsdiagnostik (staging).
Bei manchen leukämischen Lymphomen kann die Diagnose u. U. schon aus dem Blut gestellt werden. In jedem Fall sollte bei Verfügbarkeit von vitalem Tumormaterial eine zytogenetische Analyse durchgeführt werden.
Immunphänotypisierung mittels Durchflusszytometrie
Als Ausgangsmaterial für die durchflusszytometrische Immunphänotypisierung eignen sich peripheres Blut, Knochenmark, Pleuraerguss oder Aszites, wenn sie einen Lymphombefall aufweisen. Prinzipiell ist dies auch anhand von unfixiertem Lymphknotengewebe problemlos möglich, jedoch meist in der Praxis ungebräuchlich, da exstirpiertes Lymphknotengewebe in der Regel sofort fixiert wird, wonach eine immunhistochemische Färbung beim Pathologen erfolgt. Bei leukämischen Lymphomen (z. B. chronische lymphatische Leukämie, M. Waldenström, Sézary-Syndrom, leukämische Formen von: Immunozytom, Follikuläres Lymphom, Mantelzelllymphom) oder aleukämischen Lymphomen mit regelhaftem Knochenmarkbefall (z. B. Multiples Myelom) kann die Immunphänotypisierung aus Blut bzw. Knochenmark erfolgen. Mittels Immunphänotypisierung lassen sich Lymphome der T-Reihe der T-Zell-Reihe oder NK-Zell-Reihe unterscheiden. Zudem ist eine gewisse Zuordnung zum Reifungs- und Differenzierungsgrad möglich. In manchen Fällen lässt sich die Diagnose sogar schon durchflusszytometrisch stellen (Haarzellleukämie). Die meisten Lymphome weisen ein relativ charakteristisches Immunmarkerprofil auf. Jedoch gibt es in einem erheblichen Teil der Fälle auch atypische Antigen-Muster.
Zytogenetik und Molekulargenetik
Während bei der Lymphom-Diagnostik früher die rein histopathologische, das heißt morphologische Charakterisierung ganz im Vordergrund stand (später ergänzt durch die Immunhistochemie und Immunphänotypisierung), gewinnen in den letzten Jahren genetische Untersuchungen immer größere Bedeutung. Genetische Aberrationen bei NHLs sind zum Teil prognostisch oder differenzialdiagnostisch relevant und beeinflussen damit Therapieentscheidungen. Eine umfassende Lymphomdiagnostik sollte daher insbesondere bei Erstdiagnose eine genetische Analyse einschließen.
Die zytogenetische Untersuchung erfordert vitales, teilungsfähiges Tumorgewebe. Dies kann zum Beispiel unfixiertes Lymphknotengewebe oder Knochenmark-Aspirat sein. Die molekulargenetische Diagnostik bei NHL baut häufig (aber nicht notwendigerweise) auf dem Befund der zytogenetischen Diagnostik auf. Auch für die molekulargenetische Diagnostik muss das Untersuchungsmaterial unfixiert sein.
Häufige zytogenetische und molekulare Aberrationen bei Non-Hodgkin-Lymphomen

Einzelne ausgewählte Lymphom-Entitäten
Chronische lymphatische Leukämie (CLL)
Die Diagnose einer CLL kann meist schon durchflusszytometrisch aufgrund des typischen Immunphänotyps (CD5+/CD10-/CD19+/CD23+) gestellt werden. Eine genetische Untersuchung ist insbesondere bei jüngeren Patienten sinnvoll, da dadurch einzelne Hochrisiko-Patienten identifiziert werden können. Als Hochrisiko-Merkmal gelten in erster Linie die TP53-Aberrationen, die zytogenetisch als Deletion (del(17p13)) in Erscheinung treten können. Der Verlust des langen Arms von Chromosom 11 (11q-) gilt ebenfalls als prognostisch ungünstig, während die del(13q) oder Trisomie 12 als isolierte Aberrationen eine eher günstige Prognose anzeigen.Follikuläres Lymphom (FL)
Das follikuläre Lymphom wird entsprechend seinem Blastenanteil und Wachstumsmuster (follikulär versus diffus) in 3 Grade eingeteilt. Ein Grad 3 FL entspricht dabei einem hochmalignen NHL. Genetisch ist in der überwiegenden Mehrheit der Fälle die Translokation t(14;18) mit BCL2-IGH-Fusion nachweisbar. Der Nachweis ist zytogenetisch (+ FISH) und im überwiegenden Teil der Fälle auch molekulargenetisch mittels PCR möglich. Falls sich die Translokation auch molekulargenetisch nachweisen lässt können anhand dieses molekularen Markers Verlaufsuntersuchungen auf minimale Resterkrankung (MRD) durchgeführt werden. Durchflusszytometrisch zeigt sich meist ein CD5-/CD10+/CD19+/CD20+/CD23-/sIg+ Immunphänotyp. Histopathologisch sind BCL2 und BCL6 typischerweise positiv.Mantelzelllymphom (MZL)
Durchflusszytometrisch zeigt das Mantelzelllymphom einen CD5+/CD10-/CD19+/CD23- Immunphänotyp. Immunhistochemisch ist Cyclin D1 (CCND1) typischerweise positiv.
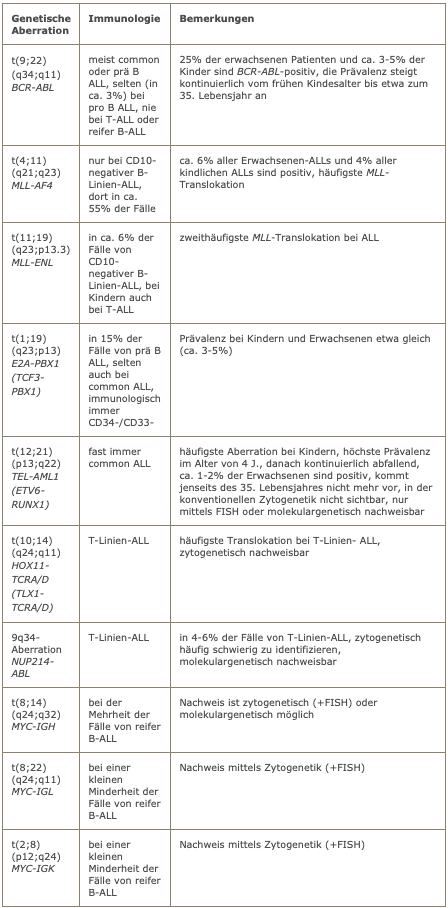
Die Translokation t(11;14) findet sich typischerweise beim Mantelzell-lymphom aber auch bei anderen NHLs.
Genetisch weisen mehr als 90% der Fälle die Translokation t(11;14) mit CCND1-IGH-Fusion auf. Der Nachweis ist gut zytogenetisch zu führen. Molekulargenetisch ist die Fusion mittels PCR nur in einem der Fälle nachweisbar, da die Chromosomenbruchpunkte weit verstreut liegen. Trotzdem sollte eine molekulargenetische Untersuchung erfolgen, wenn eine t(11;14) zytogenetisch nachweisbar ist, da sich auf diese Weise evtl. ein molekularer Marker für quantitative Verlaufsuntersuchungen identifizieren lässt.
Burkitt-Lymphom (BL)
Die differenzialdiagnostische Abgrenzung des Burkitt-Lymphoms zum Diffus-großzelligen B-NHL (DLBCL) ist gelegentlich schwierig. Große Bedeutung kommt der Morphologie und der genetischen Analyse zu. Zytogenetisch zeigen sich fast immer Translokationen des MYC-Gens auf dem langen Arm von Chromosom 8 (8q24) in die Nähe der Immunglobulin-Gene IGH (Schwerketten-Locus, 14q32), IGK (Leichtkettenlocus kappa, 2p12) oder IGL (Leichtkettenlocus lambda, 22q11).
Knochenmarkinfiltration durch ein Burkitt-Lymphom. Die Blasten zeigen zahlreiche Zytoplasmavakuolen und stark basophiles Zytoplasma.
In etwa 75-85% der Fälle von BL liegt eine Translokation t(8;14)(q24;q32) vor, die sowohl zytogenetisch, als auch molekulargenetisch nachgewiesen werden kann. In etwa 5% der Fälle zeigt sich zytogenetisch eine Translokation t(2;8)(p12;q24) und in etwa 10% eine Translokation t(8;22)(q24;q11). Alle drei Translokationen kommen allerdings auch bei anderen NHL-Entitäten vor (DLBCL, selten Multiples Myelom) und beweisen daher alleine kein BL. Zytologisch imponieren die BL-Blasten meist in Form einer „FAB L3-Morphologie“.
Diffus-großzelliges B-Zell-Lymphom (DLBCL)
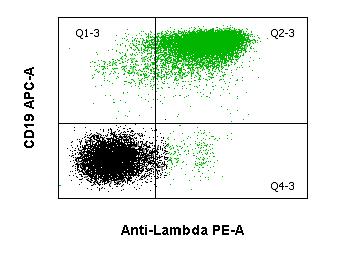
Das diffus-großzellige B-Zell-Lymphom ist ein Überbegriff für eine Gruppe von hochmalignen B-Zell-Lymphomen mit diffusem Wachstumsmuster. Die DLBCL bilden eine biologisch heterogene Gruppe. Morphologisch werden im wesentlichen die zentroblastische, immunoblastische und die anaplastische Variante unterschieden. Durchflusszytometrisch sind B-Zellantigene (CD19, CD20, CD79a) nachweisbar, sowie meist auch Oberflächen-Immunglobulin (sIg) ggf. mit Leichtketten-Restriktion. CD10 ist in einem variablen Prozentsatz exprimiert. Genetisch sind die am häufigsten betroffenen Loci 3q27 (BCL6), 18q21 (BCL2) und 8q24 (MYC) und der Immunglobulin-Schwerkettenlocus (IGH).Haarzellleukämie (HCL)
Die Haarzellleukämie ist ein seltenes niedrigmalignes B-NHL. Bei der Knochenmarkuntersuchung kann häufig nur Knochenmarkblut aspiriert werden und morphologisch typische Haarzellen finden sich nicht immer im peripheren Blut.
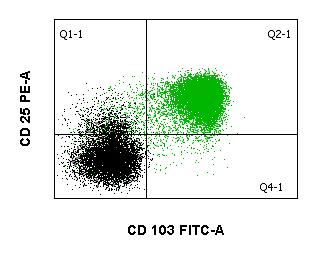
Haarzelle im peripheren Blut
Die Diagnose wird in Verbindung mit typischen klinischen Befunden in der Regel immunologisch, z. B. mittels Durchflusszytometrie gestellt, da die HCL durch die relativ spezifische Expression des CD103-Antigens gekennzeichnet ist. Die Antigene CD19, CD20, CD22, CD11c, CD25, und FMC7 sind ebenfalls exprimiert, während CD5, CD10 und CD23 typischerweise fehlen. Genetisch ist bei typischer HCL meist die BRAF V600E-Mutation, die auch bei einer Reihe von soliden Tumoren zu finden ist, nachweisbar. Bei Verdacht auf HCL sollte neben den Durchflusszytometrie auch eine molekulargenetische Untersuchung auf BRAF V600E Mutation erfolgen.
Multiples Myelom (MM)
Das Multiple Myelom ist eine klinisch und auch diagnostisch vielgestaltige Erkrankung. Durchflusszytometrisch sind plasmazelluläre Antigene (z. B. CD38 und CD 138) nachweisbar.Typischerweise liegt ein CD19-/CD79a+/CD38+/CD138+ Immunphänotyp vor. Aberrante Antigen-Expressionen (CD117, CD20, CD52, CD10) werden gelegentlich beobachtet. Knochenmarkmorphologisch lässt sich die Erkrankung meist eindeutig diagnostizieren.
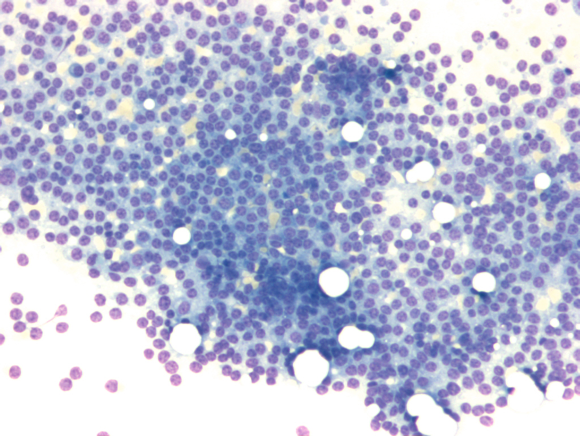
Hochgradige Knochenmarkinfiltration durch ein Multiples Myelom
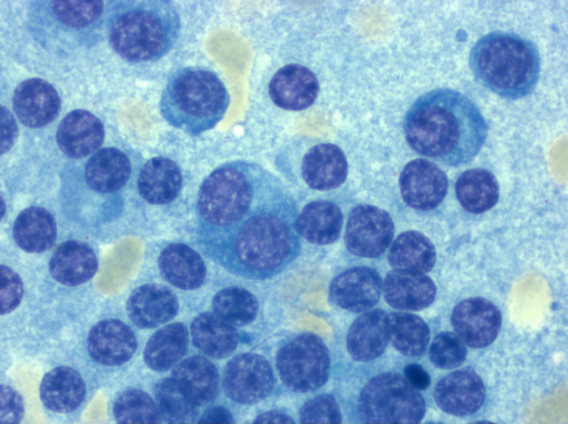
Ausschnittsvergrößerung
Die genetische Analyse des MM war lange Zeit dadurch erschwert, dass sich die Myelom-Zellen in Kultur nur sehr langsam oder gar nicht teilen, so dass eine zytogenetische Analyse häufig nicht möglich war.
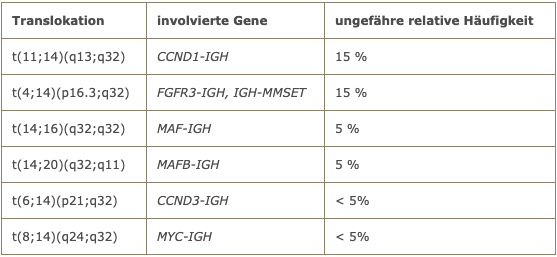
Im Labor Berlin ist ein spezielles Kulturmedium in Gebrauch, mit dem sich eine deutlich verbesserte Rate an auswertbaren Metaphasen bei Myelomzellen in Kultur erzielen lässt. In etwas mehr als der Hälfte der Fälle liegen Chromosomentranslokationen unter Beteiligung des Immunglobulin-Schwerkettenlocus (IGH) vor. Die häufigsten Chromosomentranslokationen beim Multiplen Myelom sind in der folgenden Tabelle aufgeführt.
Chromosomentranslokationen beim Multiplen Myelom
Prolymphozytenleukämie (PLL)
Die Prolymphozytenleukämie kann einen B-Zell- oder einen T-Zell-Immunphänotyp aufweisen. Morphologisch zeigt die B-PLL zum großen Teil unreife mittelgroße Prolymphozyten mit prominenten Nucleoli. Immunphänotypisch sind B-Zell-Antigene exprimiert (CD19/CD22/CD79a+b/FMC7) und eine Leichtkettenrestriktion ist nachweisbar. CD5 und CD23 sind im Unterschied zur CLL nur in einer Minderheit der Fälle nachweisbar. Genetisch zeigen sich häufig komplexe Karyotypen. Die T-PLL hat einen CD2+/CD3+/CD7+/CD52+ Phänotyp. Meist zeigt sich eine CD4+/CD8- Expression, gelegentlich sind aber CD4 und CD8 koexprimiert. Zytogenetisch sind Aberrationen der T-Zell-Rezeptorgene typisch.Sézary-Syndrom
Ein Sézary-Syndrom, d.h. ein leukämisiertes kutanes T-Zell-Lymphom liegt vor, wenn morphologisch typische Sézary-Zellen im peripheren Blut und durchflusszytometrisch eine neoplastische T-Zell-Population nachweisbar sind. Nach der WHO-Klassifikation 2008 sind für die Diagnose mindestens 1000/µl Sézary-Zellen und/oder ein CD4+/CD8+-Verhältnis von mindestens 10:1 und/oder der Verlust mehrerer T-Zell-Antigene (z.B. CD7) erforderlich. Auch bei im Blut zirkulierenden Sézary-Zellen lässt sich meist keine Knochenmarkinfiltration durch das zugrundeliegende T-Zell-Lymphom nachweisen. Typischerweise liegt ein CD2+/CD3+/CD5+/CD7-/CD4+ Immunphänotyp vor. CD8+ positive Fälle sind selten. Morphologisch zeigen Sézary-Zellen im Unterschied zu normalen Lymphozyten stark wechselnde, z. T. bizarre Kernformen. Genetisch scheint das Sézary-Syndrom heterogen und ist bisher wenig charakterisiert. -
Bei der PNH liegt eine erworbene Mutation im PIGA-Gen auf Xp22.1 vor. Daraus resultiert eine Störung der Verankerung von Glykoproteinen in der Zellmembran (der sogenannte GPI-Anker ist gestört). Zu den davon betroffenen Proteinen gehören auch die Proteine CD55 und CD59, die eine komplementinhibierende Funktion haben. Infolge des Fehlens von CD55 und CD59 kommt es bei PNH-Patienten zu einer chronischen oder anfallsartigen Komplementaktivierung. Leitsymptome der PNH sind die Coombs-negative Hämolyse, Thromboseneigung (häufig mit atypischer Lokalisation), periphere Zytopenie einer oder mehrerer Reihen und ggf. auch eine renale Funktionseinschränkung (durch Mikrothromben). Die Symptome der PNH-Patienten hängen entscheidend von der Größe des PNH-Klons ab. Nicht selten tritt die PNH im Kontext anderer hämatologischer Erkrankungen auf – häufig mit der Aplastischen Anämie, seltener bzw. weniger ausgeprägt mit Niedrigrisiko-Myelodysplastischen Syndromen.
Durchflusszytometrie
Der diagnostische „Goldstandard“ für die Diagnosestellung einer PNH ist die durchflusszytometrische Untersuchung des peripheren Blutes auf Expression GPI-verankerter Proteine. Die Leitlinien der DGHO (Stand: März 2012) verlangen die Untersuchung von mindestens 2 verschiedenen Markern (GPI-verankerte Proteine oder GPI-Anker selbst). Mindestens Granulozyten und Erythrozyten sollten untersucht werden. Der Anteil des PNH-Klons kann am besten anhand des Anteils GPI-defizienter Granulozyten abgeschätzt werden, da dieser nicht durch Hämolyse oder Transfusionen beeinflusst wird.
Wichtig ist die Probenlogistik: die eingesandte Probe muss möglichst frisch, d. h. innerhalb 48 Stunden analysiert werden. Probenabnahmen am Freitag oder vor Feiertagen sind daher zu vermeiden. Bei Transportzeiten > 24 Stunden sollte die Probe gekühlt werden (+1 bis +10 oC).
Indikationen für die Diagnostik
- Coombs-negative Hämolyse
- Thrombosen, wenn mindestens eines der folgenden Kriterien erfüllt ist:
- Atypische Lokalisation (Sinusvenenthrombose, Budd-Chiari, etc.)
- Thrombosen in Verbindung mit unklarer Zytopenie
- Thrombosen unklarer Ursache, arterielle Thrombosen
- Verdacht auf aplastische Anämie
- Verdacht auf myelodysplastisches Syndrom
- Rezidiverende Schmerzzustände bei gleichzeitig vorhandenen Zeichen einer Hämolyse




Kontakt
-
Labor Berlin – Charité Vivantes GmbH
Sylter Straße 2
13353 BerlinTel.: +49 (30) 40 50 26-800
Fax: +49 (30) 40 50 26-615 -
Campus Charité Mitte (CCM)
Tel.: +49 (30) 450-513023
Fax: +49 (30) 450-513933 -
Campus Virchow Klinikum (CVK)
Morphologie
Tel.: +49 (30) 40 50 26-498
Fax: +49 (30) 40 50 26-615Immunphänotypisierung
Tel.: +49 (30) 40 50 26-497
Fax: +49 (30) 40 50 26-615Tumorgenetik
Tel.: +49 (30) 40 50 26-493
Fax: +49 (30) 40 50 26-618Tumorzytogenetik
Tel.: +49 (30) 450-569145
Fax: +49 (30) 450-569996
Präanalytische Hinweise und Downloads
-
Antikoagulanz für alle zytologischen Untersuchungen ist grundsätzlich EDTA. In Ausnahmefällen geht auch Citrat. Heparin ist dagegen ungünstig und sollte nicht verwendet werden.
Als Untersuchungsmaterial sind geeignet:
- Knochenmarkaspirat (5 ml)
- peripheres Blut (Routine-EDTA-Blutröhrchen)
- Ergussmaterial (Aszites, Pleuraerguss, Perikarderguss u.a.)
- Liquor
Es sollte darauf geachtet werden, dass die Transportzeit nicht mehr als 24 Stunden beträgt und dass das Material vor Wochenenden spätestens bis Freitag, 14.00 Uhr im Labor eintrifft. Grundsätzlich sollte Material mit einem Express-Service (Übernacht-Service) verschickt werden, um zu lange Transportzeiten zu vermeiden.
-
Durchflusszytometrie
Antigoagulans für alle durchflusszytometrischen Untersuchungen ist grundsätzlich EDTA. Heparin ist weniger gut geeignet und sollte nur dann verwendet werden, wenn nichts anderes verfügbar ist (z.B. versehentliche Abnahme in einer falschen Spritze bei einer Knochenmarkpunktion).
Als Untersuchungsmaterial sind geeignet:
- Knochenmarkaspirat (5 – 10 ml)
- peripheres Blut ( 5 – 10 ml)
- Ergussmaterial (Aszites, Pleuraerguss, Perikarderguss u.a.), wenn möglich 10 ml
- Liquor
PNH-Diagnostik erfolgt aus peripherem Blut.
Der Probenversand sollte unbedingt mit einem Express-Service (Übernacht-Service) erfolgen, um die Transportzeiten so gering wie möglich zu halten. Bei Eingang im Labor sollten die Proben nicht älter als 24 Stunden sein. Vor Wochenenden ist darauf zu achten, dass die Probe spätestens Freitagmittag, um 12.00 Uhr im Labor eintrifft.
-
Antikoagulans für alle molekulargenetischen Untersuchungen ist grundsätzlich EDTA. In Ausnahmefällen geht auch Zitrat. Heparin ist dagegen ungünstig und sollte nur dann verwendet werden wenn nichts anderes verfügbar ist (z. B. versehentliche Abnahme in einer falschen Spritze bei einer Knochenmarkpunktion).
Als Untersuchungsmaterial sind im Prinzip möglich:
- Knochenmarkaspirat (5 ml)
- Peripheres Blut (10 ml)
- Erguss-Material (Aszites, Pleuraerguss)
- Liquor
- Lymphknoten- oder Tumorbiopsie (unfixiert !)
Bei Einsendung von Tumormaterial oder Lymphknoten wird um vorherige telefonische Rücksprache gebeten. Die molekulargenetische Diagnostik aus Liquor hat meist nur dann Aussicht auf Erfolg, wenn dieser sehr zellreich ist. Auch hier wird eine vorherige telefonische Rücksprache empfohlen.
Der Probenversand sollte insbesondere bei zeitkritischen Proben (akute Leukämie) per Eilpost erfolgen. Der Versand sollte wenn möglich nicht an einem Freitag erfolgen.
Anforderungsscheine und Downloads
Leistungen für Privatpatienten, selbstzahlende Patienten oder laborärztliche Wahlleistungen werden durch LABOR BERLIN direkt mit dem betreffenden Kostenträger abgerechnet, sofern mit dem Einsender nicht etwas anderes vereinbart ist. Hierzu wird der Einsender die notwendigen Daten der Patienten an LABOR BERLIN weiterleiten und sicherstellen, dass die Patienten über die mögliche Weiterleitung von Laboraufträgen an LABOR BERLIN und die damit verbundenen organisatorischen Maßnahmen, einschließlich der Abrechnung durch eine privatärztliche Verrechnungsstelle, in der gesetzlich vorgeschriebenen Weise informiert werden und hierin einwilligen. Die rechtlichen Vorgaben in Bezug auf die freie Arztwahl werden dabei berücksichtigt. Wir weisen darauf hin, dass die Veranlassung externer laborärztlicher Wahlleistungen nach den Regelungen des Krankenhausentgeltgesetzes (KHEntgG) durch den Einsender einzelfallbezogen und konkret durch die betreffenden Wahlärzte zu erfolgen hat.
-
Anforderungsschein Hämatologische Spezialdiagnostik (Standort CVK)
pdf -
Anforderungsschein Hämatologische Spezialdiagnostik
pdf -
Einwilligungserklärung Liquid Biopsy
pdf -
Anforderungsschein Hämatologie Zytologie (Standort CVK)
pdf -
Anforderungsschein Hämatologie (Standort CCM)
pdf -
Einverständniserklärung Tumorzytogenetik und Tumorgenetik
pdf -
Anforderungsschein Hämatologie Kombinationsschein Zytologie & Molekulargenetik (Standort CVK)
pdf -
Anforderungsschein DPD Diagnostik
pdf